 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sirius Huang | -- | 3381 | 2022-11-03 01:30:51 |
Video Upload Options
In crystallography, direct methods is a set of techniques used for structure determination using diffraction data and a priori information. It is a solution to the crystallographic phase problem, where phase information is lost during a diffraction measurement. Direct methods provides a method of estimating the phase information by establishing statistical relationships between the recorded amplitude information and phases of strong reflections.
1. Background
1.1. Phase Problem
In electron diffraction, a diffraction pattern is produced by the interaction of the electron beam and the crystal potential. The real space and reciprocal space information about a crystal structure can be related through the Fourier transform relationships shown below, where [math]\displaystyle{ f(\textbf{r}) }[/math] is in real space and corresponds to the crystal potential, and [math]\displaystyle{ F(\textbf{k}) }[/math] is its Fourier transform in reciprocal space. The vectors [math]\displaystyle{ \textbf{r} }[/math] and [math]\displaystyle{ \textbf{k} }[/math] are position vectors in real and reciprocal space, respectively.
- [math]\displaystyle{ f(\textbf{r}) = \int_{-\infty}^{\infty} F(\textbf{k}) e^{2\pi i\textbf{k}\cdot\textbf{r}} dk }[/math]
- [math]\displaystyle{ F(\textbf{k}) = \int_{-\infty}^{\infty} f(\textbf{r}) e^{-2\pi i\textbf{k}\cdot\textbf{r}} dr }[/math]
[math]\displaystyle{ F(\textbf{k}) }[/math], also known as the structure factor, is the Fourier transform of a three-dimensional periodic function (i.e. the periodic crystal potential), and it defines the intensity measured during a diffraction experiment. [math]\displaystyle{ F(\textbf{k}) }[/math] can also be written in a polar form [math]\displaystyle{ F(\textbf{g}) }[/math], where [math]\displaystyle{ \textbf{g} }[/math] is a specific reflection in reciprocal space. [math]\displaystyle{ F(\textbf{g}) }[/math] has an amplitude term (i.e. [math]\displaystyle{ |F(\textbf{g})| }[/math]) and a phase term (i.e. [math]\displaystyle{ e^{i\phi_g} }[/math]). The phase term contains the position information in this form.
- [math]\displaystyle{ F(\textbf{g}) = |F(\textbf{g})| e^{i\phi_g} }[/math]
During a diffraction experiment, the intensity of the reflections are measured as [math]\displaystyle{ I(\textbf{g}) }[/math]:
- [math]\displaystyle{ I(\textbf{g}) = |F(\textbf{g})|^2 }[/math]
This is a straightforward method of obtaining the amplitude term of the structure factor. However, the phase term, which contains position information from the crystal potential, is lost.
Analogously, for electron diffraction performed in a transmission electron microscope, the exit wave function of the electron beam from the crystal in real and reciprocal space can be written respectively as:
- [math]\displaystyle{ \psi(\textbf{r}) = a(\textbf{r}) e^{-i\phi(\textbf{r})} }[/math]
- [math]\displaystyle{ \Psi(\textbf{u}) = A(\textbf{u}) e^{-i\phi(\textbf{u})} }[/math]
Where [math]\displaystyle{ a(\textbf{r}) }[/math] and [math]\displaystyle{ A(\textbf{u}) }[/math] are amplitude terms, the exponential terms are phase terms, and [math]\displaystyle{ \textbf{u} }[/math] is a reciprocal space vector. When a diffraction pattern is measured, only the intensities can be extracted. A measurement obtains a statistical average of the moduli:
- [math]\displaystyle{ I(\textbf{r}) = \langle |\psi(\textbf{r})|^2 \rangle = \langle |a(\textbf{r})|^2 \rangle }[/math]
- [math]\displaystyle{ I(\textbf{u}) = \langle |\Psi(\textbf{u})|^2 \rangle = \langle |A(\textbf{u})|^2 \rangle }[/math]
Here, it is also clear that the phase terms are lost upon measurement in an electron diffraction experiment. This is referred to as the crystallographic phase problem.
1.2. History
In 1952, David Sayre introduced the Sayre equation, a construct that related the known phases of certain diffracted beams to estimate the unknown phase of another diffracted beam.[1] In the same issue of Acta Crystallographica, Cochran and Zachariasen also independently derived relationships between the signs of different structure factors.[2][3] Later advancements were done by other scientists, including Hauptman and Karle, leading to the awarding of the Nobel Prize in Chemistry (1985) to Hauptman and Karle for their development of direct methods for the determination of crystal structures.[4]
1.3. Comparison to X-Ray Direct Methods
The majority of direct methods was developed for X-ray diffraction. However, electron diffraction has advantages in several applications. Electron diffraction is a powerful technique for analyzing and characterizing nano- and micron-sized particles, molecules, and proteins. While electron diffraction is often dynamical and more complex to understand compared to X-ray diffraction, which is usually kinematical, there are specific cases (detailed later) that have sufficient conditions for applying direct methods for structure determination.
2. Theory
2.1. Unitary Sayre Equation
The Sayre equation was developed under certain assumptions taken from information about the crystal structure, specifically that all atoms considered are identical and there is a minimum distance between atoms.[1] Called the "Squaring Method," a key concept of the Sayre equation is that squaring the electron-density function (for X-ray diffraction) or crystal potential function (for electron diffraction) results in a function that resembles the original un-squared function of identical and resolved peaks. By doing so, it reinforces atom-like features of the crystal.
Consider the structure factor [math]\displaystyle{ F(\textbf{k}) }[/math] in the following form, where [math]\displaystyle{ f(\textbf{k}) }[/math] is the atomic scattering factor for each atom at position [math]\displaystyle{ \textbf{k} }[/math], and [math]\displaystyle{ \textbf{r} }[/math] is the position of atom [math]\displaystyle{ l }[/math]:
- [math]\displaystyle{ F(\textbf{k}) = \sum_l f(\textbf{k}) e^{2 \pi \textbf{k} \cdot \textbf{r}_{l}} }[/math]
This can be converted to the unitary structure factor [math]\displaystyle{ U(\textbf{k}) }[/math] by dividing by N (the number of atoms) and [math]\displaystyle{ f(\textbf{k}) }[/math]:
- [math]\displaystyle{ U(\textbf{k}) = \frac{1}{N} \sum_l e^{2 \pi \textbf{k} \cdot \textbf{r}_{l}} }[/math]
This can be alternatively rewritten in real and reciprocal space as:
- [math]\displaystyle{ u(\textbf{r}) = \frac{1}{N} \sum_l \delta (\textbf{r}-\textbf{r}_{l}) = N u(\textbf{r})^2 }[/math]
- [math]\displaystyle{ U(\textbf{k}) = N \sum_h U(\textbf{k-h}) U(\textbf{h}) }[/math]
This equation is a variation of the Sayre equation. Based on this equation, if the phases of [math]\displaystyle{ U(\textbf{k-h}) }[/math] and [math]\displaystyle{ U(\textbf{h}) }[/math] are known, then the phase of [math]\displaystyle{ U(\textbf{k}) }[/math] is known.
2.2. Triplet Phase Relationship
The triplet phase relationship is an equation directly relating two known phases of diffracted beams to the unknown phase of another. This relationship can be easily derived via the Sayre equation, but it may also be demonstrated through statistical relationships between the diffracted beams, as shown here.
For randomly distributed atoms, the following holds true:
- [math]\displaystyle{ N \langle U(\textbf{k-h}) U(\textbf{h}) \rangle = \frac{1}{N} \sum_l e^{2 \pi i \textbf{k} \cdot \textbf{r}_l} = U(\textbf{k}) }[/math]
Meaning that if:
- [math]\displaystyle{ U(\textbf{k}) \approx N \langle U(\textbf{k-h}) U(\textbf{h}) \rangle }[/math]
Then:
- [math]\displaystyle{ |U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})|^2 = |U(\textbf{k})|^2 + N^2 |U(\textbf{k-h})U(\textbf{h})|^2 - 2 N |U(\textbf{k}) U(\textbf{k-h}) U(\textbf{h})| \times cos(\phi(\textbf{k}) - \phi(\textbf{k-h}) - \phi(\textbf{h})) }[/math]
In the above equation, [math]\displaystyle{ \langle |U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})|^2 \rangle = 0 }[/math] and the moduli are known on the right hand side. The only unknown terms are contained in the cosine term that includes the phases. The central limit theorem can be applied here, which establishes that distributions tend to be Gaussian in form. By combining the terms of the known moduli, a distribution function can be written that is dependent on the phases:
- [math]\displaystyle{ P(U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})) \approx C e^{-|U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})|^2} }[/math]
- [math]\displaystyle{ P(U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})) \approx D e^{2 N |U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})| \times cos(\phi(\textbf{k}) - \phi(\textbf{k-h}) - \phi(\textbf{h}))} }[/math]
This distribution is known as the Cochran distribution.[5] The standard deviation for this Gaussian function scales with the reciprocal of the unitary structure factors. If they are large, then the sum in the cosine term must be:
- [math]\displaystyle{ \phi(\textbf{k}) - \phi(\textbf{k-h}) - \phi(\textbf{h}) \approx 2n\pi,~~n = 0, 1, 2... }[/math]
- [math]\displaystyle{ \phi(\textbf{k}) \approx \phi(\textbf{k-h}) - \phi(\textbf{h}) }[/math]
This is called the triplet phase relationship ([math]\displaystyle{ \Sigma_2 }[/math]). If the phases [math]\displaystyle{ \phi(\textbf{k-h}) }[/math] and [math]\displaystyle{ \phi(\textbf{h}) }[/math] are known, then the phase [math]\displaystyle{ \phi(\textbf{k}) }[/math] can be estimated.
2.3. Tangent Formula
The tangent formula was first derived in 1955 by Jerome Karle and Herbert Hauptman.[4] It related the amplitudes and phases of known diffracted beams to the unknown phase of another. Here, it is derived using the Cochran distribution.
- [math]\displaystyle{ \prod_h P(U(\textbf{k}) - N U(\textbf{k-h}) U(\textbf{k})) \approx 2 N C e^{\sum_h |U(\textbf{k}) U(\textbf{k-h}) U(\textbf{k})| \times cos(\phi(\textbf{k}) - \phi(\textbf{k-h}) - \phi(\textbf{h}))} }[/math]
The most probable value of [math]\displaystyle{ \phi(\textbf{h}) }[/math] can be found by taking the derivative of the above equation, which gives a variant of the tangent formula:[6]
- [math]\displaystyle{ tan(\varphi(\textbf{k})) \approx \frac{ \sum_h |U(\textbf{k}) - NU(\textbf{k-h}) U(\textbf{h}) | sin \phi(\textbf{k-h}) + \phi(\textbf{h}) }{\sum_h |U(\textbf{k}) - NU(\textbf{k-h}) U(\textbf{h}) | cos \phi(\textbf{k-h}) + \phi(\textbf{h})} }[/math]
3. Practical Considerations
The basis behind the phase problem is that phase information is more important than amplitude information when recovering an image. This is because the phase term of the structure factor contains the positions. However, the phase information does not need to be retrieved completely accurately. Often even with errors in the phases, a complete structure determination is possible. Likewise, amplitude errors will not severely impact the accuracy of the structure determination.
3.1. Sufficient Conditions
In order to apply direct methods to a set of data for successful structure determination, there must be reasonable sufficient conditions satisfied by the experimental conditions or sample properties. Outlined here are several cases.[6]
- Kinematical Diffraction
One of the reasons direct methods was originally developed for analyzing X-ray diffraction is because almost all X-ray diffraction is kinematical. While most electron diffraction is dynamical, which is more difficult to interpret, there are instances in which mostly kinematical scattering intensities can be measured. One specific example is surface diffraction in plan view orientation. When analyzing the surface of a sample in plan view, the sample is often tilted off a zone axis in order to isolate the diffracted beams of the surface from those of the bulk. Achieving kinematical conditions is difficult in most cases—it requires very thin samples to minimize dynamical diffraction.
- Statistical Kinematical Diffraction
Even though most cases of electron diffraction are dynamical, it is still possible to achieve scattering that is statistically kinematical in nature. This is what enables the analysis of amorphous and biological materials, where dynamical scattering from random phases add up to be nearly kinematical. Furthermore, as explained earlier, it is not critical to retrieve phase information completely accurately. Errors in the phase information are tolerable.
Recalling the Cochran distribution and considering a logarithm of that distribution:
- [math]\displaystyle{ D(\phi(\textbf{k})-\phi(\textbf{k-h})-\phi(\textbf{h})) = A(\textbf{k,h}) \sqrt[]{I(\textbf{k}) I(\textbf{k-h}) I(\textbf{h})} \times cos (\phi(\textbf{k})-\phi(\textbf{k-h})-\phi(\textbf{h})) }[/math]
- [math]\displaystyle{ D(\phi(\textbf{k})-\phi(\textbf{k-h})-\phi(\textbf{h})) = B(\textbf{k,h}) cos(\phi(\textbf{k})-\phi(\textbf{k-h})-\phi(\textbf{h})) }[/math]
In the above distribution, [math]\displaystyle{ A(\textbf{k}) }[/math] contains normalization terms, [math]\displaystyle{ I(\textbf{k}) }[/math] terms are the experimental intensities, and [math]\displaystyle{ B(\textbf{k,h}) }[/math] contains both of these for simplicity. Here, the most probable phases will maximize the function [math]\displaystyle{ D(\textbf{k,h}) }[/math]. If the intensities are sufficiently high and the sum in the cosine term remains [math]\displaystyle{ \approx 0 }[/math], then [math]\displaystyle{ B(\textbf{k,h}) }[/math] will also be large, thereby maximizing [math]\displaystyle{ D(\textbf{k,h}) }[/math]. With a narrow distribution such as this, the scattering data will be statistically within the realm of kinematical consideration.
- Intensity Mapping
Consider two scattered beams with different intensities. The magnitude of their intensities will then have to be related to the amplitude of their corresponding scattering factors by the relationship:
- [math]\displaystyle{ I(\textbf{k}) \gt I(\textbf{k'})~~ iff~|F(\textbf{k})| \gt |F(\textbf{k'})| }[/math]
Let [math]\displaystyle{ T(\textbf{k} }[/math]) be a function that relates the intensity to the phase for the same beam, where [math]\displaystyle{ N(\textbf{k}) }[/math] contains normalization terms:
- [math]\displaystyle{ T(\textbf{k}) = e^{i\phi(\textbf{k})} \sqrt[]{I(\textbf{k})}/N(\textbf{k}) }[/math]
Then, the distribution of [math]\displaystyle{ T(\textbf{k}) }[/math] values will be directly related to the values of [math]\displaystyle{ F(\textbf{k}) }[/math]. That is, when the product [math]\displaystyle{ |F(\textbf{k})F(\textbf{k-h})F(\textbf{h})| }[/math] is large or small, [math]\displaystyle{ |T(\textbf{k})T(\textbf{k-h})T(\textbf{h})| }[/math] will also be large and small. So, the observed intensities can be used to reasonably estimate the phases for diffracted beams. The observed intensity can be related to the structure factor more formally using the Blackman formula.[7]
Other cases to consider for intensity mapping are specific diffraction experiments, including powder diffraction and precession electron diffraction. Specifically, precession electron diffraction produces a quasi-kinematical diffraction pattern that can be used adequately in direct methods.
- Dominated Scattering
In some cases, scattering from a sample can be dominated by one type of atom. Therefore, the exit wave from the sample will also be dominated by that atom type. For example, the exit wave and intensity of a sample dominated by channeling can be written in reciprocal space in the form:
- [math]\displaystyle{ \Psi(\textbf{k}) = A(\textbf{k}) \sum_l e^{2 \pi i \textbf{k} \cdot \textbf{r}_l} }[/math]
- [math]\displaystyle{ I(\textbf{k}) = |A(\textbf{k})|^2 \bigg| \sum_l e^{2 \pi i \textbf{k} \cdot \textbf{r}_l} \bigg|^2 }[/math]
[math]\displaystyle{ A(\textbf{k}) }[/math] is the Fourier transform of [math]\displaystyle{ a(\textbf{r}) }[/math], which is complex and represents the shape of an atom, given by the channeling states (e.g. 1s, 2s, etc.). [math]\displaystyle{ A(\textbf{k}) }[/math] is real in reciprocal space and complex in the object plane. If [math]\displaystyle{ B(\textbf{k}) }[/math], a conjugate symmetric function, is substituted for [math]\displaystyle{ A(\textbf{k}) }[/math], then it is feasible to retrieve atom-like features from the object plane:
- [math]\displaystyle{ B(\textbf{k}) = S(\textbf{k}) |A(\textbf{k})|, where ~S(\textbf{k}) = \pm 1 }[/math]
In the object plane, the Fourier transform of [math]\displaystyle{ B(\textbf{k}) }[/math] will be a real and symmetric pseudoatom ([math]\displaystyle{ b(\textbf{r}) }[/math]) at the atomic column positions. [math]\displaystyle{ b(\textbf{r}) }[/math] will satisfy atomistic constraints as long as they are reasonably small and well-separated, thereby satisfying some constraints required for implementing direct methods.
4. Implementation
Direct methods is a set of routines for structure determination. In order to successfully solve for a structure, several algorithms have been developed for direct methods. A selection of these are explained below.
4.1. Gerchberg-Saxton
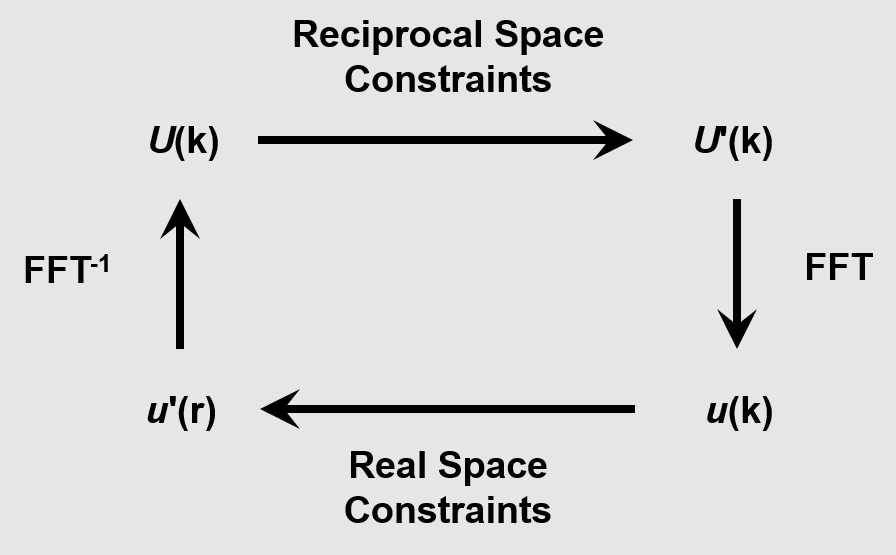
The Gerchberg-Saxton algorithm was originally developed by Gerchberg and Saxton to solve for the phase of wave functions with intensities known in both the diffraction and imaging planes.[8] However, it has been generalized for any information in real or reciprocal space. Detailed here is a generalization using electron diffraction information. As illustrated in image to the right,[6] one can successively impose real space and reciprocal constraints on an initial estimate until it converges to a feasible solution.
4.2. Constraints
Constraints can be physical or statistical. For instance, the fact that the data is produced by a scattering experiment in a transmission electron microscope imposes several constraints, including atomicity, bond lengths, symmetry, and interference. Constraints may also be statistical in origin, as shown earlier with the Cochran distribution and triplet phase relationship ([math]\displaystyle{ \Sigma_2 }[/math]).
According to Combettes, image recovery problems can be considered as a convex feasibility problem.[9] This idea was adapted by Marks et al. to the crystallographic phase problem.[10] With a feasible set approach, constraints can be considered convex (highly convergent) or non-convex (weakly convergent). Imposing these constraints with the algorithm detailed earlier can converge towards unique or non-unique solutions, depending on the convexity of the constraints.
4.3. Examples
Direct methods with electron diffraction datasets have been used to solve for a variety of structures. As mentioned earlier, surfaces are one of the cases in electron diffraction where scattering is kinematical. As such, many surface structures have been solved for by both X-ray and electron diffraction direct methods, including many of the silicon, magnesium oxide, germanium, copper, and strontium titanate surfaces.[11][12][13]
More recently, methods for automated three dimensional electron diffraction methods have been developed, such as automated diffraction tomography and rotation electron diffraction. These techniques have been used to obtain data for structure solution through direct methods and applied for zeolites, thermoelectrics, oxides, metal-organic frameworks, organic compounds, and intermetallics.[14] In some of these cases, the structures were solved in combination with X-ray diffraction data, making them complementary techniques.
In addition, some success has been found using direct methods for structure determination with the cryo-electron microscopy technique Microcrystal Electron Diffraction (MicroED).[15] MicroED has been used for a variety of materials, including crystal fragments, proteins, and enzymes.[16]
5. Software
5.1. DIRDIF
DIRDIF is a computer program for structure determination through using the Patterson function and direct methods applied to difference structure factors. It was first released by Paul Beurkens and his colleagues at the University of Nijmegen in 1999. It is written in Fortran and was most recently updated in 2008. It can be used for structures with heavy atoms, structures of molecules with partly known geometries, and for certain special case structures. Detailed information can be found at its website: http://www.xtal.science.ru.nl/dirdif/software/dirdif.html.
5.2. EDM
Electron Direct Methods is a set of programs developed at Northwestern University by Professor Laurence Marks. First released in 2004, its most recent release was version 3.1 in 2010. Written in C++, C, and Fortran 77, EDM is capable of performing image processing of high resolution electron microscopy images and diffraction patterns and direct methods. It has a standard GNU license and is free to use or modify for non-commercial purposes. It uses a feasible set approach [10] and genetic algorithm search for solving structures using direct methods, and it also has high-resolution transmission electron microscopy image simulation capabilities. More information can be found at the website: http://www.numis.northwestern.edu/edm/index.shtml.
5.3. OASIS
OASIS was first written by several scientists from the Chinese Academy of Sciences in Fortran 77. The most recent release is version 4.2 in 2012. It is a program for direct methods phasing of protein structures. The acronym OASIS stands for two of its applications: phasing One-wavelength Anomalous Scattering or Single Isomorphous Substitution protein data. It reduces the phase problem to a sign problem by locating the atomic sites of anomalous scatterers or heavy atom substitutions. More details can be found at the website: http://cryst.iphy.ac.cn/Project/IPCAS1.0/user_guide/oasis.html.
5.4. SIR
The SIR (seminvariants representation) suite of programs was developed for solving the crystal structures of small molecules. SIR is updated and released frequently, with the first release in 1988 and the latest release in 2014. It is capable of both ab initio and non-ab-initio direct methods. The program is written in Fortran and C++ and is free for academic use. SIR can be used for the crystal structure determination of small-to-medium-sized molecules and proteins from either X-ray or electron diffraction data. More information can be found at its website: http://www.ba.ic.cnr.it/softwareic/sir2014/.
References
- Sayre, D. (1 January 1952). "The squaring method: a new method for phase determination". Acta Crystallographica 5 (1): 60–65. doi:10.1107/S0365110X52000137. https://dx.doi.org/10.1107%2FS0365110X52000137
- Cochran, W. (1 January 1952). "A relation between the signs of structure factors". Acta Crystallographica 5 (1): 65–67. doi:10.1107/S0365110X52000149. https://dx.doi.org/10.1107%2FS0365110X52000149
- Zachariasen, W. H. (1 January 1952). "A new analytical method for solving complex crystal structures". Acta Crystallographica 5 (1): 68–73. doi:10.1107/S0365110X52000150. https://dx.doi.org/10.1107%2FS0365110X52000150
- Karle, J.; Hauptman, H. (1 August 1956). "A theory of phase determination for the four types of non-centrosymmetric space groups 1P222, 2P22, 3P12, 3P22". Acta Crystallographica 9 (8): 635–651. doi:10.1107/S0365110X56001741. https://dx.doi.org/10.1107%2FS0365110X56001741
- Cochran, W. (10 August 1955). "Relations between the phases of structure factors". Acta Crystallographica 8 (8): 473–478. doi:10.1107/S0365110X55001485. https://dx.doi.org/10.1107%2FS0365110X55001485
- Marks, L. D.; Sinkler, W. (16 September 2003). "Sufficient Conditions for Direct Methods with Swift Electrons". Microscopy and Microanalysis 9 (5): 399–410. doi:10.1017/S1431927603030332. PMID 19771696. Bibcode: 2003MiMic...9..399M. https://dx.doi.org/10.1017%2FS1431927603030332
- Blackman, M. (10 November 1939). "On the Intensities of Electron Diffraction Rings". Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences 173 (952): 68–82. doi:10.1098/rspa.1939.0129. Bibcode: 1939RSPSA.173...68B. https://dx.doi.org/10.1098%2Frspa.1939.0129
- Gerchberg, R. W.; Saxton, W. O. (29 November 1971). "A Practical Algorithm for the Determination of Phase from Image and Diffraction Plane Pictures". Optik 35 (2): 237–246.
- Combettes, P. L. (1 January 1996). The Convex Feasibility Problem in Image Recovery. 95. Elsevier. 155–270. doi:10.1016/S1076-5670(08)70157-5. ISBN 9780120147373. https://dx.doi.org/10.1016%2FS1076-5670%2808%2970157-5
- Marks, L. D.; Sinkler, W.; Landree, E. (1 July 1999). "A feasible set approach to the crystallographic phase problem". Acta Crystallographica Section A 55 (4): 601–612. doi:10.1107/S0108767398014408. PMID 10927270. https://dx.doi.org/10.1107%2FS0108767398014408
- Marks, L. D.; Bengu, E.; Collazo-Davila, C.; Grozea, D.; Landree, E.; Leslie, C.; Sinkler, W. (October 1998). "Direct Methods for Surfaces". Surface Review and Letters 05 (5): 1087–1106. doi:10.1142/S0218625X98001444. Bibcode: 1998SRL.....5.1087M. https://dx.doi.org/10.1142%2FS0218625X98001444
- Erdman, N.; Poeppelmeier, K. R.; Asta, M.; Warschkow, O.; Ellis, D. E.; Marks, L. D. (5 September 2002). "The structure and chemistry of the TiO2-rich surface of SrTiO3 (001)". Nature 419 (6902): 55–58. doi:10.1038/nature01010. PMID 12214229. Bibcode: 2002Natur.419...55E. https://dx.doi.org/10.1038%2Fnature01010
- Kienzle, Danielle M.; Marks, Laurence D. (2012). "Surface transmission electron diffraction for SrTiO3 surfaces". CrystEngComm 14 (23): 7833. doi:10.1039/c2ce25204j. https://dx.doi.org/10.1039%2Fc2ce25204j
- Yun, Y.; Zou, X.; Hovmöller, S.; Wan, W. (10 February 2015). "Three-dimensional electron diffraction as a complementary technique to powder X-ray diffraction for phase identification and structure solution of powders". IUCrJ 2 (2): 267–282. doi:10.1107/S2052252514028188. PMID 25866663. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4392419
- de la Cruz, M. J.; Hattne, J.; Shi, D.; Seidler, P.; Rodriguez, J.; Reyes, F. E.; Sawaya, M. R.; Cascio, D. et al. (13 February 2017). "Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED". Nature Methods 14 (4): 399–402. doi:10.1038/nmeth.4178. PMID 28192420. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5376236
- Nannenga, Brent L.; Gonen, Tamir (2018-02-06). "MicroED: a versatile cryoEM method for structure determination". Emerging Topics in Life Sciences 2 (1): 1–8. doi:10.1042/etls20170082. ISSN 2397-8554. PMID 30167465. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6112783

