 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hilma van der Horst | + 2504 word(s) | 2504 | 2020-11-23 06:47:44 |
Video Upload Options
Monoclonal antibody (mAb) therapy has rapidly changed the field of cancer therapy. In 1997, the CD20-targeting mAb rituximab was the first mAb to be approved by the U.S. Food and Drug Administration (FDA) for treatment of cancer. Within two decades, dozens of mAbs entered the clinic for treatment of several hematological cancers and solid tumors, and numerous more are under clinical investigation. The success of mAbs as cancer therapeutics lies in their ability to induce various cytotoxic machineries against specific targets. These cytotoxic machineries include antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC), which are all mediated via the fragment crystallizable (Fc) domain of mAbs. In this review article we will outline the novel approaches of engineering these Fc domains of mAbs to enhance their Fc-effector function and thereby their anti-tumor potency, with specific focus to summarize their (pre-) clinical status for the treatment of B-cell malignancies, including chronic lymphocytic leukemia (CLL), B-cell Non-Hodgkin lymphoma (B-NHL), and multiple myeloma (MM).
1. Introduction
Naturally, antibodies (Abs) are produced by B-cells as a polyclonal population, with high specificity for their distinct target antigen and epitope. Antibodies thereby play various important roles in our immune system. The field of therapeutic Abs commenced in 1975, when the development of the mouse hybridoma technology enabled the production of large amounts of murine monoclonal (m) Abs [1]. However, murine mAbs elicited an immunogenic response in human patients. To reduce this immunogenicity, chimeric mAbs, consisting of a constant human domain fused to a variable mouse domain, were developed [2]. The chimeric mAb rituximab targeting cluster of differentiation (CD) 20 was the first FDA-approved mAb for cancer therapy in 1997. The development of advanced design technologies such as human antibody-gene expression libraries and transgenic animals allowed the engineering of humanized (the hypervariable region of a murine antibody grafted in a human antibody) and fully human mAbs [3]. To be successfully applied in the clinic, mAbs generally require additional engineering to improve their affinity, limit any biophysical liabilities, and to increase their half-life. Currently, 30 mAbs are clinically-approved for treatment of cancer, and this number is rapidly increasing: in the last decade the number of mAbs that have entered late-stage clinical studies has been tripled [4]. The therapeutic potential of mAbs has been exploited by the development of antibody fusion products, such as bispecific antibodies or antibody drug conjugates, which take advantage of specific antigen binding properties of antibodies to precisely target cytotoxic cells or toxic agents to cancerous cells. A novel development in antibody engineering is the modification of the antibody fragment crystallizable (Fc) region in order to increase the Fc-tail mediated effector functions, including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC), to induce tumor cytotoxicity more effectively. Numerous Fc-engineered antibodies have demonstrated clinical activity or are under preclinical investigation.
In this review we will outline the novel approaches of engineering Fc-domains of mAbs to enhance their Fc-effector function and anti-tumor potency, with specific focus to their (pre-) clinical status for the treatment of B-cell malignancies, including chronic lymphocytic leukemia (CLL), B-cell Non-Hodgkin lymphoma (B-NHL), and multiple myeloma (MM). Not described in this review is the application of Fc-engineering in order to improve antibody half-life, to silence mAb effector functions in case of antibodies used as receptor agonists or antagonists or as drug delivery vehicles, and to increase the direct, not Fc-effector function mediated, anti-tumor potency of mAbs.
2. Antibody Structure
Antibodies are mono- or polymers of immunoglobulins (Ig) consisting of two identical pairs of heavy (H) and light (L) chains, which are linked through non-covalent interactions and disulfide bonds to form a Y-shaped structure [5]. All H and L light chains contain a single variable domain (VL), which also consists of hypervariable regions. The combination of the (hyper) variable regions of the H and L chains determines the antigen specificity and affinity of an antibody. The L chains contain a single constant (CL) domain to make a stable link with the H chain. The number of constant domains of H chain (CH) is dependent on the isotype of the antibody: IgA, IgD and IgG contain three (CH1-3), and IgE and IgM contain four constant domains (CH1-4) C domains. The first CH is linked with CL to the variable regions, which together form the fragment antigen binding (Fab) region. The heavily glycosylated CH2-3 or CH2-4 domains are linked to CH1 via a flexible hinge region and constitute the Fc region (Figure 1A). The engineering of this region, which is responsible for the isotype- and subclass dependent Fc-mediated effector functions of antibodies will be the main focus of this review.
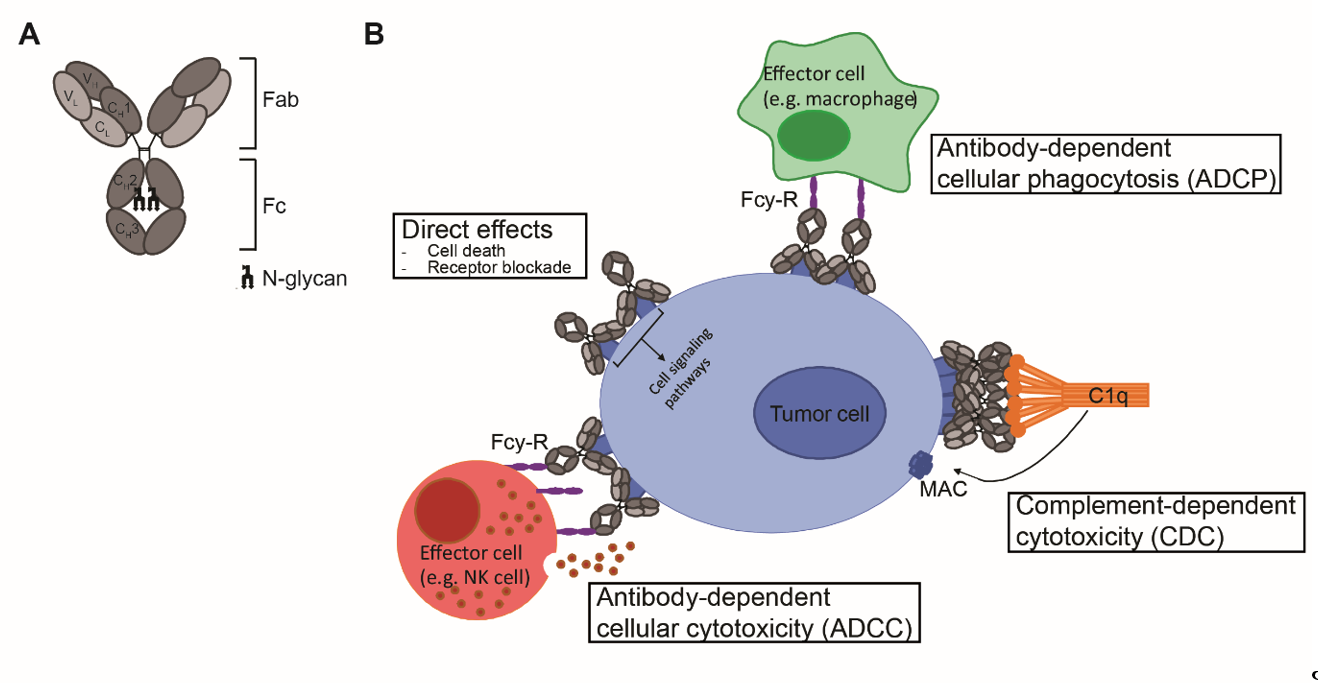
Figure 1. mAb structure and mechanisms of action. (A) Schematic representation of an IgG antibody consisting of variable (V) and constant (C) domains of the light (L; light grey) and heavy (H; dark grey) chains. The fragment-antigen binding (Fab) domain is made up of VL and CL together with VH and CH1, and the fragment crystallizable (Fc) region consist of CH2 and CH3, with an N-glycan attached to CH2. (B) Mechanisms of action of IgG1 mAbs consist of direct effects, including cell death and receptor blockade, and indirect effects, including ADCC, ADCP and CDC. ADCC and ADCP are mediated via binding to FcγR and cell death occurs via release of cytotoxic granules and via internalization and degradation of target, respectively. CDC is mediated via binding to complement protein C1q and cell death occurs via formation of the membrane attack complex (MAC), which consists of complement proteins C5b, C6, C7 and C8 and various copies of C9, and generates pores in the membrane.
3. Fc-effector functions
Upon antigen engagement, IgG antibodies can induce direct anti-tumor effects via triggering the cell death signaling pathways and via blockade of essential receptor systems, as well as indirect anti-tumor effects via their Fc-mediated effector functions, by engaging other immune cells or killer mechanisms. The Fc- mediated effector functions of antibodies include Fcγ-receptor (FcγR) mediated ADCC, Fcγ-receptor (FcγR) mediated ADCP, and complement protein C1q-mediated CDC, and have been shown to be crucial for the therapeutic efficacy of most clinically-approved antibodies (Figure 1B). Among the four IgG subclasses, IgG1 and IgG3 induce the strongest Fc-effector functions [6]. However, since IgG1 has the longest half-life and is more stable than IgG3 [7], most therapeutic antibodies with Fc-mediated functions are of IgG1 isotype.
4. Fc-engineering to enhance Fc-effector functions
The hinge and the proximal CH2 regions of the Fc-tail are considered critical for Fc-interaction with FcγRs and C1q. The interface of these regions contains the binding sites while the structural conformation of the CH2 domain allows engagement of C1q or FcγR. In addition, the CH2 domains are post-translationally modified by asparagine(N)297-linked glycosylation, and glycosylation and the specific glycan composition contribute to the stability and the dynamics of the CH2 domains [8][9][10]. Glycan components include core units of N-acetylglucosamine (GlcNAc) and mannose, with additional variations in galactose, bisecting GlcNAc, fucose and sialic acid.
Detailed understanding of Fc-interactions with C1q and FcγR opened up opportunities to modulate C1q- and FcγR- binding by Fc-engineering. In order to enhance ADCC, ADCP and CDC, studies have employed site-directed mutagenesis (sequence variations), Fc-glycosylation modification (glycoengineering), and avidity modulation, which are illustrated in figure 2.
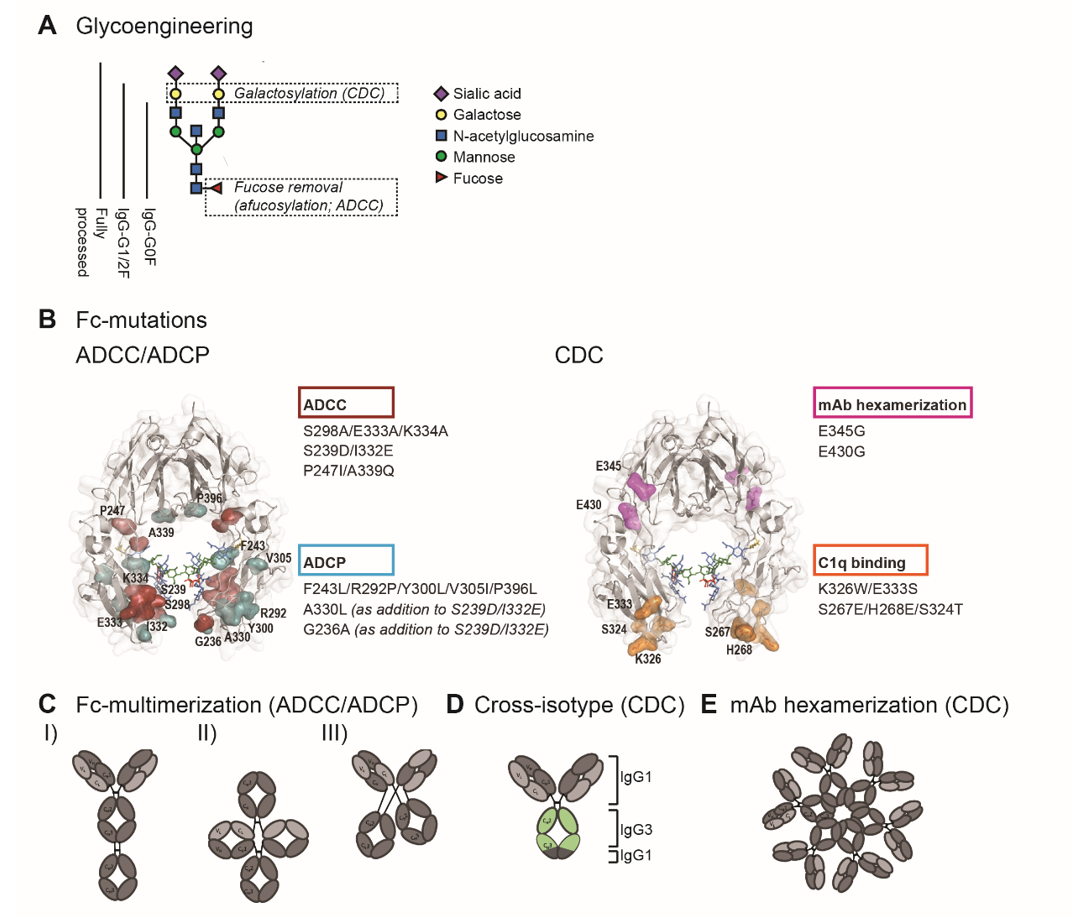
Figure 2. Fc-engineering strategies to enhance Fc-effector functions. (A) The biantennary complex glycan structure of the most frequent variants for the N-glycan of therapeutic IgG (G0F, G1F, G2F and fully processed). Glycoengineering strategies focus on galactosylation for enhanced CDC and afucosylation for enhanced ADCC. (B) ADCC/ADCP (left) and CDC (right) enhancing point mutations depicted in the crystal structure of the Fc region (PDB:3DO3). The ADCC-enhancing mutations S298A/E333A/K334A, S239D/I3332E and P247I/A339Q are depicted in magenta, the ADCP-enhancing mutations F234L/R292P/Y300L/V305I/P396L, A330L and G236A are shown in blue. A330L and G236A were added to the ADCC-enhancing mutations S239D/I332E to decrease the mutation-related increase in affinity to the inhibitory receptor FcγRIIb. The CDC-enhancing mutations E345G and E430G, which induce Fc:Fc interactions and subsequent mAb hexamerization (see E), are shown in orange and K326W/E333S and S267E/H268E/S324T, which enhance C1q binding affinity, are shown in pink. (C) Schematic representation of three Fc-multimerization strategies which are aimed to enhance ADCC and ADCP. (D) Cross-isotype antibody generated by replacing the CH2 and part of the CH3 domains of an IgG1 antibody with the corresponding regions of an IgG3 antibody, to increase the CDC response. (E) Schematic representation of mAb hexamerization which facilitates C1q binding and enhances CDC, and can occur upon antigen binding by IgG1 antibodies naturally or when harboring the E345G or E430g mutations.
5. Generation of Fc-engineered mAbs
Although the hybridoma technology revolutionized the field of therapeutic antibodies, most mAbs that are currently approved for therapeutic use are generated by mammalian expression systems, which allow higher antibody yields and preserve post-translational modifications, generating a higher quality mAb product. Mammalian expression systems often use the variable regions derived from the hybridoma or phage display technologies. The sequence of the desired region is cloned into the appropriate expression vector and subsequently transfected into the expression system. Currently available mammalian expression systems include various Chinese hamster ovary (CHO) cell lines, mouse myeloma (NS0), and mouse hybridoma (Sp2/0) cell lines. In addition, several human expression systems are available, including embryonic kidney (HEK293), amniotic (CAP), a hybrid of HEK293 and lymphoma (HKB-11) and embryonic retina (PER.C6). The human expression systems, however, provide transient expression and are therefore only suitable for preclinical purposes.
To create glycoengineered antibodies in order to develop antibodies with improved ADCC activity, several modified mammalian expression systems were developed. Double knockout of the enzyme α1,6-fucosyltransferase 8 (FUT8), which catalyzes the transfer of fucose from GDP-fucose to N-acetylglucosamine (GlcNAc), in CHO cell lines resulted in the production of afucosylated antibodies [11]. Alternatively, CHO cells were engineered to express β(1,4)–N–acetylglucosaminyltransferase III (GnTIII). IgGs produced by mammalian cells have very low or no bisecting GlcNAc, in contrast to IgGs present in human serum, and increasing the number of bisecting GlcNAc improved ADCC levels [12][13]).
6. Clinical experience with Fc-engineered mAbs for B-cell malignancies
Although various strategies can enhance ADCC, ADCP or CDC effector function, they do not uniformly increase these effector functions when applied to different antigens, since antigen binding also affects the C1q and FcγR binding via structural allostery [14][15]. In addition, there is a partial overlap in the Fc-binding sites for C1q and FcγR [6]. Therefore modulating the Fc-tail to enhance ADCC/ADCP can negatively influence CDC, and vice versa. It is thus recommended to evaluate Fc-effector function-enhancing strategies for each target individually.
In B-cell malignancies a wide variety of disease-associated targets are available, including various lineage specific surface molecules. An overview of Fc-engineered mAbs in (pre-)clinical development for treatment of B-cell malignancies is shown in Table 1.
Table 1. Fc-engineered mAbs with enhanced mAb effector function in (pre-) clinical development for CLL, B-NHL and MM.
|
Target |
Iso-type |
Chimeric/ human(ized) |
Fc-engineering strategy |
Enhanced effector function |
Additional mAb-engineering |
Clinical stage; NCT of recruiting clinical trials |
Major indication(s) |
|
|
Obinutuzumab (GA101;Gazyva) |
CD20, type II |
IgG2 |
Humanized |
Afucosylation |
ADCC |
Modified elbow-hinge |
FDA-approved |
FL and CLL |
|
Ublituximab (LFB-R603, EMAB-6) |
CD20, type I |
IgG1 |
Chimeric |
Low fucose |
ADCC |
|
Phase 2/3 1 |
CLL and B-NHL |
|
Ocaratuzumab (AME-133v, LY2469298) |
CD20, type I |
IgG1 |
Humanized |
Mutations P247I/A339Q |
ADCC |
Antigen binding affinity optimized |
Discontinued |
|
|
PRO131921 (RhuMAb; v114) |
CD20, type I |
IgG1 |
Humanized |
Mutations (na) |
ADCC and CDC |
|
Discontinued |
|
|
Ocrelizumab |
CD20 |
IgG1 |
Humanized |
Mutations (na) |
ADCC |
|
Discontinued in hematology, approved for MS |
MS |
|
CD20 double engineered |
CD20 |
IgG1 |
|
Afucosylation+ mutations S267E/H268F/S324T/G236A/I332E |
ADCC and CDC |
|
Preclinical |
|
|
CD20 |
IgG1/ IgG3 |
|
Afucosylation + mixed IgG1/IgG3 isotype |
ADCC and CDC |
|
Preclinical |
|
|
|
BI 836826 |
CD37 |
IgG1 |
Chimeric |
Mutations S239D/I332E |
ADCC |
|
Discontinued |
|
|
DuoHexaBody-CD37 (GEN3009) |
CD37 |
IgG1 |
Human |
Mutation E430G (HexaBody) |
CDC |
Dual-epitope targeting |
Phase 1 2 |
B-NHL |
|
Ianalumab (VAY736; B-1239) |
BAFF-R |
IgG1 |
Human |
Afucosylation |
ADCC |
|
Phase 1 3 |
CLL |
|
Inebilizumab (MEDI-551) |
CD19 |
IgG1κ |
Humanized |
Afucosylation |
ADCC |
|
Phase 1/2 |
B-cell malignancies |
|
MDX-1342 |
CD19 |
|
Human |
Afucosylation |
ADCC and ADCP |
|
Phase 1 halted |
|
|
Tafasitamab (MOR208, XmAb5574) |
CD19 |
IgG1 |
Humanized |
Mutations S239D/I332E |
ADCC and ADCP |
Antigen binding affinity optimized |
Priority review granted by FDA |
CLL and DLBCL |
|
HexaBody-CD38 (GEN3014) |
CD38 |
IgG1 |
Human |
Mutation E430G (HexaBody) |
CDC |
|
Preclinical |
B-NHL and MM |
|
anti-CD38 SIFbody |
CD38 |
|
|
Fc multimerization |
ADCC and CDC |
|
Preclinical |
MM |
|
XmAb5592 |
HM1.24 |
IgG1 |
Humanized |
Mutations S239D/I332E |
|
|
|
MM |
|
ICAM-1 |
IgG1 |
Human |
Mutations S239D/I332E |
ADCC and ADCP |
|
Preclinical |
MM |
|
|
SEA-BCMA |
BCMA |
IgG1 |
Humanized |
Afucosylation |
ADCC and ADCP |
|
Phase 1 4 |
MM |
1 NCT03828448; NCT04149821; NCT03801525; NCT02535286; NCT03379051; NCT04016805; NCT02793583
2 NCT04358458
3 NCT03400176
4 NCT03582033
7. Conclusion and future perspective
In this review we have illustrated various strategies to enhance Fc-mediated effector functions and we have summarized their clinical application in B-CLL, B-NHL or MM. Fc-mediated effector functions are being enhanced to increase their anti-tumor potency or when a specific effector function is beneficial, i.e. improving ADCC induction for combination therapy with lenalidomide or in patients with low affinity FcγR-polymorphisms. In addition, Fc-engineering can be applied to antibodies which depend on antibody clustering or FcγR-mediated antibody crosslinking for agonism of receptors, such as antibodies targeting the costimulatory protein CD40 or antibodies targeting death receptors 4 or 5, and thus benefit from similar strategies as discussed here [16][17][18].
Generally, mAbs that are Fc-engineered to improve their effector functions are capable of enhancing in vitro and in vivo anti-tumor potency compared to their parental unmodified mAb. Various Fc-engineered mAbs also demonstrated clinical efficacy, and are already approved for clinical use. However, other Fc-engineered mAbs demonstrated toxicity in clinical trials or failed to induce significant clinical efficacy, and were discontinued for development. Increasing the clinical success of Fc-engineered mAbs requires more empirical in vitro/in vivo screening to determine the most favorable Fc-engineering strategy or combination of strategies . In addition, understanding i) the specific contribution of ADCC, ADCP and CDC to clinical efficacy of mAbs in hematological malignancies and ii) the exact clinical effect of the different Fc-engineering strategies could allow to develop Fc-engineering strategies customized to specific target and disease. A step forward in understanding the specific contribution of mAb effector functions is the recent development of Fc-engineering strategies that enhance CDC specifically, such as the HexaBody technology. Until recently, clinically evaluated mAbs were mostly Fc-engineered to enhance ADCC function. Hence, evaluating Fc-engineered mAbs with enhanced CDC and potentially comparing them to mAbs with enhanced ADCC could provide crucial information regarding the contribution of mAb effector functions to clinical efficacy and toxicity, and the results of such clinical trials are highly anticipated.
To conclude, our advanced knowledge of Fc-structure and Fc-mediated effector function has enabled the clinical development of Fc-engineered mAbs. Expanding our clinical experience with these Fc-engineered mAbs will provide valuable information that could allow the development of antibodies with tailor-made effector function.
References
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495-7.
- Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A. 1984;81(21):6851-5.
- Vaughan TJ, Osbourn JK, Tempest PR. Human antibodies by design. Nature biotechnology. 1998;16(6):535-9.
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs. 2020;12(1):1703531.
- Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446-8.
- de Taeye SW, Rispens T, Vidarsson G. The Ligands for Human IgG and Their Effector Functions. Antibodies (Basel, Switzerland). 2019;8(2).
- Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol. 2016;40:62-9.
- Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. Journal of molecular biology. 2003;325(5):979-89.
- Bowden TA, Baruah K, Coles CH, Harvey DJ, Yu X, Song BD, et al. Chemical and structural analysis of an antibody folding intermediate trapped during glycan biosynthesis. Journal of the American Chemical Society. 2012;134(42):17554-63.
- Jefferis R. Isotype and glycoform selection for antibody therapeutics. Archives of biochemistry and biophysics. 2012;526(2):159-66.
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnology and Bioengineering. 2004;87(5):614-22.
- Davies J, Jiang L, Pan L-Z, LaBarre MJ, Anderson D, Reff M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FCγRIII. Biotechnology and Bioengineering. 2001;74(4):288-94.
- Umaña P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nature biotechnology. 1999;17(2):176-80.
- Yogo R, Yamaguchi Y, Watanabe H, Yagi H, Satoh T, Nakanishi M, et al. The Fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Scientific reports. 2019;9(1):11957.
- Zhao J, Nussinov R, Ma B. Antigen binding allosterically promotes Fc receptor recognition. MAbs. 2019;11(1):58-74.
- Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcγ receptor engagement. Proc Natl Acad Sci U S A. 2012;109(27):10966-71.
- White AL, Dou L, Chan HTC, Field VL, Mockridge CI, Moss K, et al. Fcγ Receptor Dependency of Agonistic CD40 Antibody in Lymphoma Therapy Can Be Overcome through Antibody Multimerization. The Journal of Immunology. 2014;193(4):1828-35.
- Overdijk MB, Strumane K, Beurskens FJ, Ortiz Buijsse A, Vermot-Desroches C, Vuillermoz BS, et al. Dual epitope targeting and enhanced hexamerization by DR5 antibodies as a novel approach to induce potent anti-tumor activity through DR5 agonism. Molecular cancer therapeutics. 2020.


