 +1 credit
+1 credit
Video Upload Options
GENCODE is a scientific project in genome research and part of the ENCODE (ENCyclopedia Of DNA Elements) scale-up project. The GENCODE consortium was initially formed as part of the pilot phase of the ENCODE project to identify and map all protein-coding genes within the ENCODE regions (approx. 1% of Human genome). Given the initial success of the project, GENCODE now aims to build an “Encyclopedia of genes and genes variants” by identifying all gene features in the human and mouse genome using a combination of computational analysis, manual annotation, and experimental validation, and annotating all evidence-based gene features in the entire human genome at a high accuracy. The result will be a set of annotations including all protein-coding loci with alternatively transcribed variants, non-coding loci with transcript evidence, and pseudogenes.
1. Current Progress
GENCODE is currently progressing towards its goals in Phase 2 of the project, which are:
- To continue improving the coverage and accuracy of the GENCODE human gene set by enhancing and extending the annotation of all evidence-based gene features in the human genome at a high accuracy, including protein-coding loci with alternatively splices variants, non-coding loci and pseudogenes.[1]
- To create a mouse GENCODE gene set that includes protein-coding regions with associated alternative splice variants, non-coding loci which have transcript evidence, and pseudogenes.[1]
The most recent release of the Human geneset annotations is Gencode 32, with a freeze date of September 2019. This release utilises the latest GRCh38 human reference genome assembly.[2]
The latest release for the mouse geneset annotations is Gencode M23, also with a freeze date of September 2019.[2]
Since September 2009, GENCODE has been the human gene set used by the Ensembl project and each new GENCODE release corresponds to an Ensembl release.
2. History
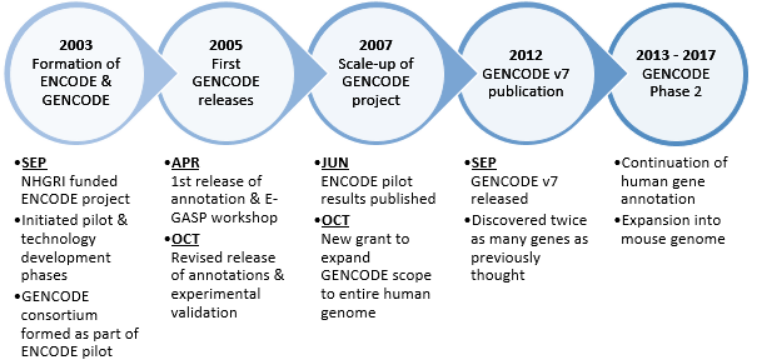
2003 September
The National Human Genome Research Institute (NHGRI) launched a public research consortium named ENCODE, the Encyclopedia Of DNA Elements, in September 2003, to carry out a project to identify all functional elements in the human genome sequence. The project was designed with three phases - Pilot, Technology development and Production phase.[3] The pilot stage of the ENCODE project aimed to investigate in great depth, computationally and experimentally, 44 regions totaling 30 Mb of sequence representing approximately 1% of the human genome. As part of this stage, the GENCODE consortium was formed to identify and map all protein-coding genes within the ENCODE regions.[4] It was envisaged that the results of the first two phases will be used to determine the best path forward for analysing the remaining 99% of the human genome in a cost-effective and comprehensive production phase.[3]
2005 April
The first release of the annotation of the 44 ENCODE regions was frozen on 29 April 2005 and was used in the first ENCODE Genome Annotation Assessment Project (E-GASP) workshop.[4] GENCODE Release 1 contained 416 known loci, 26 novel (coding DNA sequence) CDS loci, 82 novel transcript loci, 78 putative loci, 104 processed pseudogenes and 66 unprocessed pseudogenes.
2005 October
A second version (release 02) was frozen on 14 October 2005, containing updates following discoveries from experimental validations using RACE and RT-PCR techniques.[4] GENCODE Release 2 contained 411 known loci, 30 novel CDS loci, 81 novel transcript loci, 83 putative loci, 104 processed pseudogenes and 66 unprocessed pseudogenes.
2007 June
The conclusions from the pilot project were published in June 2007.[5] The findings highlighted the success of the pilot project to create a feasible platform and new technologies to characterise functional elements in the human genome, which paves the way for opening research into genome-wide studies.
2007 October
After a successful pilot phase on 1% of the genome, the Wellcome Trust Sanger Institute was awarded a grant from the US National Human Genome Research Institute (NHGRI) to carry out a scale-up of the GENCODE project for integrated annotation of gene features.[6] This new funding was part of NHGRI's endeavour to scale-up the ENCODE Project to a production phase on the entire genome along with additional pilot-scale studies.
2012 September
In September 2012, The GENCODE consortium published a major paper discussing the results from a major release – GENCODE Release 7, which was frozen in December 2011. The GENCODE 7 release used a combination of manual gene annotation from the Human and Vertebrate Analysis and Annotation (HAVANA) group and full new release (Ensembl release 62) of the automatic gene annotation from Ensembl. At the time of release, GENCODE Release 7 had the most comprehensive annotation of long noncoding RNA (lncRNA) loci publicly available with the predominant transcript form consisting of two exons.[7]
2013 - 2017
Having been involved in successfully delivering the definitive annotation of functional elements in the human genome, the GENCODE group were awarded a second grant in 2013 in order to continue their human genome annotation work and expand GENCODE to include annotation of the mouse genome.[8] It is envisaged that the mouse annotation data will allow comparative studies between the human and mouse genomes, to improve annotation quality in both genomes.
3. Key Participants
The key participants of the GENCODE project have remained relatively consistent throughout its various phases, with the Wellcome Trust Sanger Institute now leading the overall efforts of the project.
A summary of key participating institutions of each phase is listed below:
| GENCODE Phase 2 (Current)[9] | GENCODE Scale-up Phase[6] | GENCODE Pilot Phase[10] | |
|---|---|---|---|
| Wellcome Trust Sanger Institute, Cambridge, UK | Wellcome Trust Sanger Institute, Cambridge, UK | Wellcome Trust Sanger Institute, Cambridge, UK
|
|
| Centre de Regulació Genòmica (CRG), Barcelona, Catalonia, Spain | Centre de Regulació Genòmica (CRG), Barcelona, Catalonia, Spain | Institut Municipal d'Investigació Mèdica (IMIM), Barcelona, Catalonia, Spain | |
| The University of Lausanne, Switzerland | The University of Lausanne, Switzerland | University of Geneva, Switzerland | |
| University of California, Santa Cruz (UCSC), California, USA | University of California (UCSC), Santa Cruz, USA | Washington University (WashU), St Louis, USA | |
| Massachusetts Institute of Technology (MIT), Boston, USA | Massachusetts Institute of Technology (MIT), Boston, USA | University of California, Berkeley, USA | |
| Yale University (Yale), New Haven, USA | Yale University (Yale), New Haven, USA | European Bioinformatics Institute, Hinxton, UK | |
| Spanish National Cancer Research Centre (CNIO), Madrid, Spain | Spanish National Cancer Research Centre (CNIO), Madrid, Spain | ||
| Washington University (WashU), St Louis, USA |
4. Key Statistics
Since its inception, GENCODE has released 20 versions of the Human gene set annotations (excluding minor updates).
The key summary statistics of the most recent GENCODE Human gene set annotation (Release 20, April 2014 freeze, Ensembl 76), which is the first version that utilises the latest version of the Human Genome Assembly (GRCh38), is shown below:[11]
| Categories | Total | Categories | Total |
|---|---|---|---|
| Total No of Genes | 58,688 | Total No of Transcripts | 194,334 |
| Protein-coding genes | 19,942 | Protein-coding transcripts | 79,460 |
| Long non-coding RNA genes | 14,470 | - full length protein-coding: | 54,447 |
| Small non-coding RNA genes | 9,519 | - partial length protein-coding: | 25,013 |
| Pseudogenes | 14,363 | Nonsense mediated decay transcripts | 13,229 |
| - processed pseudogenes: | 10,736 | Long non-coding RNA loci transcripts | 24,489 |
| - unprocessed pseudogenes: | 3,202 | ||
| - unitary pseudogenes: | 171 | ||
| - polymorphic pseudogenes: | 26 | ||
| - pseudogenes: | 2 | ||
| Immunoglobulin/T-cell receptor gene segments | 618 | Total No of distinct translations | 59,575 |
| - protein coding segments: | 392 | Genes that have more than one distinct translations | 13,579 |
| - pseudogenes: | 226 |
Refer to the GENCODE Statistics README and GENCODE biotypes page for more details on the classification of the above gene set.
Through advancements in sequencing technologies (such as RT-PCR-seq), increased coverage from manual annotations (HAVANA group), and improvements to automatic annotation algorithms using Ensembl, the accuracy and completeness of GENCODE annotations have been continuously refined through its iteration of releases.
A comparison of key statistics from 3 major GENCODE releases is shown below.[11] It is evident that although the coverage, in terms of total number of genes discovered, is steady increasing, the number of protein-coding genes has actually decreased. This is mostly attributed to new experimental evidence obtained using Cap Analysis Gene Expression (CAGE) clusters, annotated PolyA sites, and peptide hits.[7]
- Version 7 (December 2010 freeze, GRCh37) - Ensembl 62
- Version 10 (July 2011 freeze, GRCh37) - Ensembl 65
- Version 20 (April 2014 freeze, GRCh38) - Ensembl 76
-
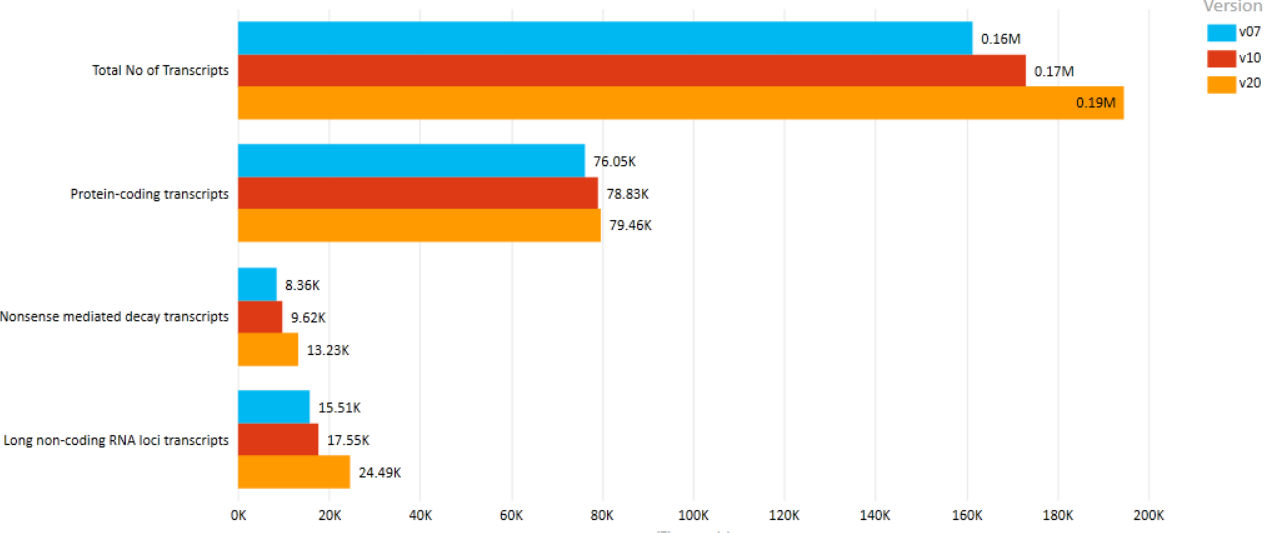
Comparison of GENCODE Human versions (Transcripts). https://handwiki.org/wiki/index.php?curid=1524080
-
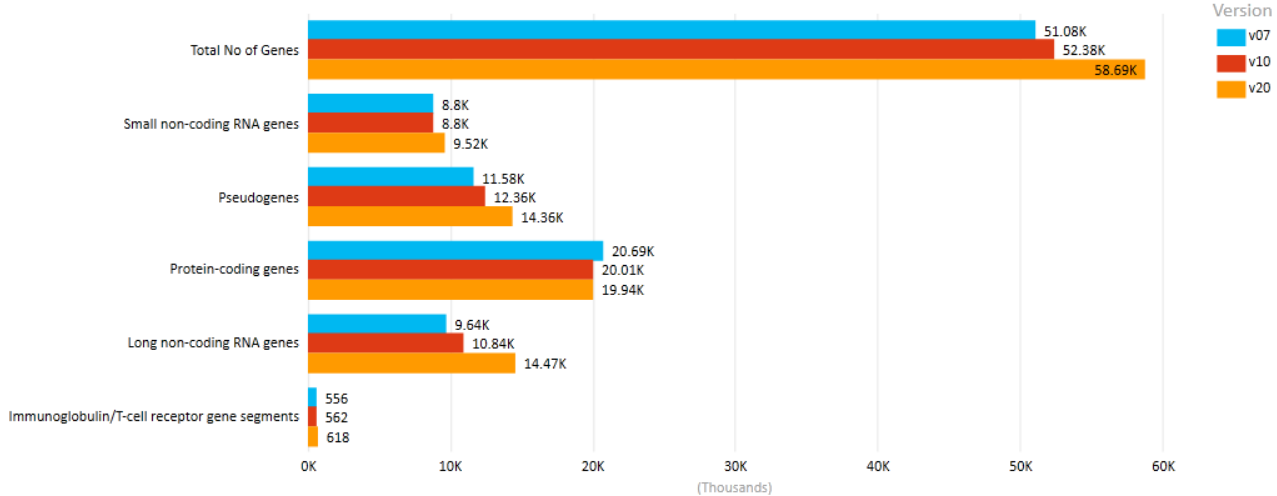
Comparison of GENCODE Human versions (Genes). https://handwiki.org/wiki/index.php?curid=1409032
-
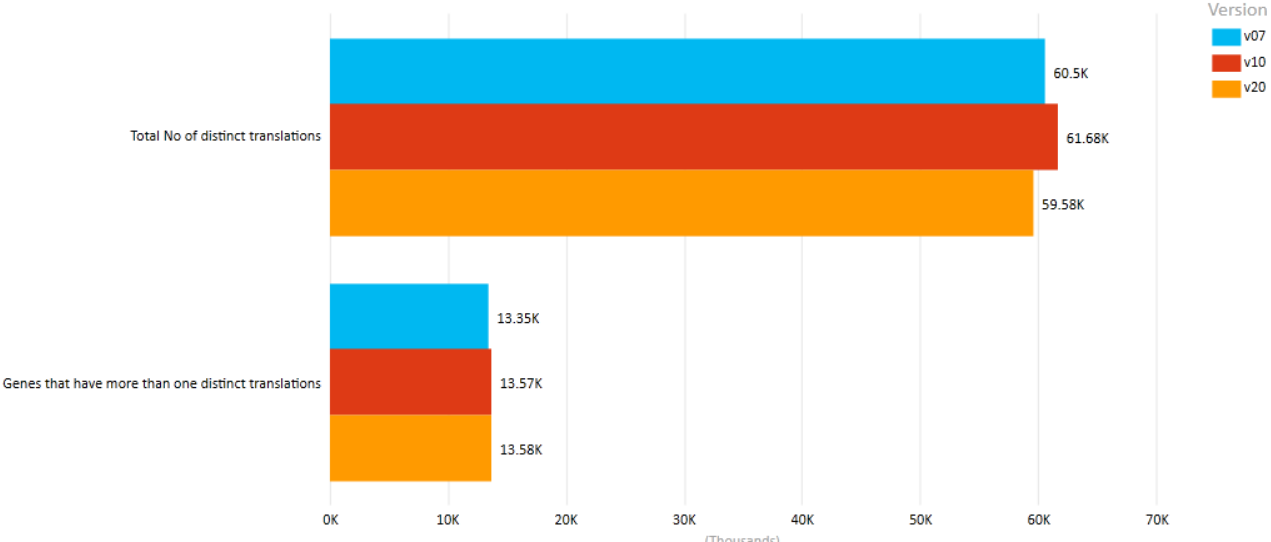
Comparison of GENCODE Human versions (Translations). https://handwiki.org/wiki/index.php?curid=1531543
5. Methodology

The general process to create an annotation for GENCODE involves manual curation, different computational analysis and targeted experimental approaches. Putative loci can be verified by wet-lab experiments and computational predictions are analysed manually.[1] Currently, to ensure a set of annotation covers the complete genome rather than just the regions that have been manually annotated, a merged data set is created using manual annotations from HAVANA, together with automatic annotations from the Ensembl automatically annotated gene set. This process also adds unique full-length CDS predictions from the Ensembl protein coding set into manually annotated genes, to provide the most complete and up-to-date annotation of the genome possible.[12]
5.1. Automatic Annotation (Ensembl)
Ensembl transcripts are products of the Ensembl automatic gene annotation system (a collection of gene annotation pipelines), termed the Ensembl gene build. All Ensembl transcripts are based on experimental evidence and thus the automated pipeline relies on the mRNAs and protein sequences deposited into public databases from the scientific community.[13] Moreover, Protein levels 1 and 2 from UniProt, untranslated regions (UTRs), long intergenic noncoding RNA (lincRNA) genes (annotated using a combination of cDNA sequences and regulatory data from the Ensembl project), short non-coding RNAs (annotated using the Ensembl ncRNA pipelines) are included.[7]
5.2. Manual Annotation (HAVANA group)
The main approach to manual gene annotation is to annotate transcripts aligned to the genome and take the genomic sequences as the reference rather than the cDNAs. The finished genomic sequence is analyzed using a modified Ensembl pipeline, and BLAST results of cDNAs/ESTs and proteins, along with various ab initio predictions, can be analyzed manually in the annotation browser tool Otterlace. Thus, more alternative spliced variants can be predicted compared with cDNA annotation. Moreover, genomic annotation produces a more comprehensive analysis of pseudogenes.[7] There are several analysis groups in the GENCODE consortium that run pipelines that aid the manual annotators in producing models in unannotated regions, and to identify potential missed or incorrect manual annotation, including completely missing loci, missing alternative isoforms, incorrect splice sites and incorrect biotypes. These are fed back to the manual annotators using the AnnoTrack tracking system.[14] Some of these pipelines use data from other ENCODE subgroups including RNASeq data, histone modification and CAGE and Ditag data. RNAseq data is an important new source of evidence, but generating complete gene models from it is a difficult problem. As part of GENCODE, a competition was run to assess the quality of predictions produced by various RNAseq prediction pipelines (Refer to RGASP below). To confirm uncertain models, GENCODE also has an experimental validation pipeline using RNA sequencing and RACE [12]
5.3. Ensembl/HAVANA Gene Merge process
During the merge process, all HAVANA and Ensembl transcripts models are compared, first by clustering overlapped coding exons on a same strand, and then by pairwise comparisons of each exon in a cluster of transcripts. The module used to merge the gene set is HavanaAdder. Additional steps are required prior to running the HavanaAdder code (e.g. Ensembl health-checking system and queries against CCDS gene set and Ensembl´s cDNA alignments). If annotation described in external data sets is missing from the manual set, then this is stored in the AnnoTrack system to be reviewed.[7]
5.4. Assessing quality
For GENCODE 7, transcript models are assigned a high or low level of support based on a new method developed to score the quality of transcripts. This method relies on mRNA and EST alignments supplied by UCSC and Ensembl. The mRNA and EST alignments are compared to the GENCODE transcripts, and the transcripts are scored according to the alignment over its full length. A summary of support levels for each chromosome in GENCODE Release 7 is shown in the figure on the right. The annotations are partitioned into those produced by the automated process, manual method and the merged annotations, where both processes result in the same annotation.[7]
5.5. General Methods Used for GENCODE 7
Amplification, sequencing, mapping and validation exon–exon junction
Double-stranded cDNA of eight human tissues (brain, heart, kidney, testis, liver, spleen, lung, and skeletal muscle) were generated with a cDNA amplification, and the purified DNA was directly used to generate a sequencing library with the ‘‘Genomic DNA sample prep kit’’ (Illumina). This library was subsequently sequenced on an Illumina Genome Analyzer 2 platform. Then, reads (35 or 75 nt) were mapped on to the reference human genome (hg19) and the predicted spliced amplicons with Bowtie software. Only uniquely mapping reads with no mismatch were considered to validate a splice site (transcript). Splice junctions were validated if a minimum of 10 reads with the following characteristics spanned the predicted splice junctions. For 35- and 75-nt-long reads, it required at least 4 and 8 nt on each side of the breakpoints (i.e., on each targeted exon), respectively.[7]
Comparison of RefSeq, UCSC, AceView, and GENCODE transcripts
Transcripts belonging to four different data sets (GENCODE, RefSeq, UCSC, and AceView) were compared to assess to which extent these data sets overlap. Releases compared were GENCODE 7, RefSeq and UCSC Genes freeze July 2011, and AceView 2010 release. The overlaps between different data set combinations were graphically represented as three-way Venn diagrams using the Vennerable R package and edited manually.[7]
PhyloCSF analysis
PhyloCSF was used to identify potential novel coding genes in RNA-seq transcript models based on evolutionary signatures. For each transcript model generated from the Illumina HBM data using either Exonerate or Scripture, a mammalian alignment was generated by extracting the alignment of each exon from UCSC’s vertebrate alignments (which includes 33 placental mammals).[7]
APPRIS (CNIO)
APPRIS is a system that deploys a range of computational methods to provide value to the annotations of the human genome. APPRIS also selects one of the CDS for each gene as the principal isoform. Moreover, it defines the principal variant by combining protein structural and functional information and information from the conservation of related species. The APPRIS server has been used in the context of the scale up of the ENCODE project to annotate the Human genome but APPRIS is being used for other species (e.g. mouse, rat and zebrafish).[15] The pipeline is made up of separate modules that combine protein structure and function information and evolutionary evidence. Each module has been implemented as a separate web service.
6. Usage/Access
The current GENCODE Human gene set version (GENCODE Release 20) includes annotation files (in GTF and GFF3 formats), FASTA files and METADATA files associated with the GENCODE annotation on all genomic regions (reference-chromosomes/patches/scaffolds/haplotypes). The annotation data is referred on reference chromosomes and stored in separated files which include: Gene annotation, PolyA features annotated by HAVANA, (Retrotransposed) pseudogenes predicted by the Yale & UCSC pipelines, but not by HAVANA, long non-coding RNAs, and tRNA structures predicted by tRNA-Scan. Some examples of the lines in the GTF format are shown below:

The columns within the GENCODE GTF file formats are described below.
Format description of GENCODE GTF file. TAB-separated standard GTF columns
| Column number | Content | Values/format |
|---|---|---|
| 1 | chromosome name | chr{1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,X,Y,M} |
| 2 | annotation source | {ENSEMBL,HAVANA} |
| 3 | feature-type | {gene,transcript,exon,CDS,UTR,start_codon,stop_codon,Selenocysteine} |
| 4 | genomic start location | integer-value (1-based) |
| 5 | genomic end location | integer-value |
| 6 | score (not used) | . |
| 7 | genomic strand | {+,-} |
| 8 | genomic phase (for CDS features) | {0,1,2,.} |
| 9 | additional information as key-value pairs | See explanation in table below. |
Description of key-value pairs in 9th column of the GENCODE GTF file (format: key "value")
| Key name | Value format |
|---|---|
| gene_id | ENSGXXXXXXXXXXX |
| transcript_id | ENSTXXXXXXXXXXX |
| gene_type | list of biotypes |
| gene_status | {KNOWN,NOVEL,PUTATIVE} |
| gene_name | string |
| transcript_type | list of biotypes |
| transcript_status | {KNOWN,NOVEL,PUTATIVE} |
| transcript_name | string |
| exon_number | indicates the biological position of the exon in the transcript |
| exon_id | ENSEXXXXXXXXXXX |
| level |
|
6.1. Level Definition
Each gene in the GENCODE data set are classified into three levels according to their type of annotation:
Level 1 (verified loci): Includes transcripts that have been manually annotated and experimentally validated by RT-PCR-seq, and pseudogenes that have been validated by three different methodologies.[7]
Level 2 (manually annotated loci): Highlights transcripts that have been manually annotated by HAVANA only, and also includes transcripts that have been merged with models produced by the Ensembl automatic pipeline.[7]
Level 3 (automatically annotated loci): Indicates transcripts and pseudogene predictions resulting from Ensembl’s automated annotation pipeline.[7]
6.2. Gene/Transcript Status Definition
Genes & transcripts are assigned the status ‘‘known,’’ ‘‘novel,’’ or ‘‘putative’’ depending on their presence in other major databases and the evidence used to build their component transcripts.
Known: Represented in the HUGO Gene Nomenclature Committee (HGNC) database and RefSeq.[7]
Novel: Not currently represented in HGNC or RefSeq databases, but are well supported by either locus specific transcript evidence or evidence from a paralogous or orthologous locus.[7]
Putative: Not currently represented in HGNC or RefSeq databases, but are supported by shorter, more sparse transcript evidence.[7]
6.3 Biodalliance Genome Browser
Also, the GENCODE website contains a Genome Browser for human and mouse where you can reach any genomic region by giving the chromosome number and start-end position (e.g. 22:30,700,000..30,900,000), as well as by ENS transcript id (with/without version) , ENS gene id (with/without version) and gene name. The browser is powered by Biodalliance.
7. Challenges
7.1. Definition of a "gene"
The definition of a "gene" has never been a trivial issue, with numerous definitions and notions proposed throughout the years since the discovery of the human genome. First, genes were conceived in the 1900s as discrete units of heredity, then it was thought as the blueprint for protein synthesis, and in more recent times, it was being defined as genetic code that is transcribed into RNA. Although the definition of a gene has evolved greatly over the last century, it has remained a challenging and controversial subject for many researchers. With the advent of the ENCODE/GENCODE project, even more problematic aspects of the definition have been uncovered, including alternative splicing (where a series of exons are separated by introns), intergenic transcriptions, and the complex patterns of dispersed regulation, together with non-genic conservation and the abundance of noncoding RNA genes. As GENCODE endeavours to build an encyclopaedia of genes and gene variants, these problems presented a mounting challenge for the GENCODE project to come up with an updated notion of a gene.[16]
7.2. Pseudogenes
Pseudogenes have DNA sequences which are similar to functional protein-coding genes, however their transcripts are usually identified with a frameshift or deletion, and are generally annotated as a byproduct of protein-coding gene annotation in most genetic databases. However, recent analysis of retrotransposed pseudogenes have found some retransposed pseudogenes to be expressed and functional and to have major biological/regulatory impacts on human biology. To deal with the unknowns and complexities of pseudogenes, GENCODE has created a pseudogene ontology using a combination of automated, manual, and experimental methods to associate a variety of biological properties—such as sequence features, evolution, and potential biological functions to pseudogenes.[7]
8. Related Projects
8.1. ENCODE
The Encyclopedia Of DNA Elements (ENCODE) is a public research consortium launched by the National Human Genome Research Institute (NHGRI), in September 2003 (Pilot phase). The goal of ENCODE is to build a comprehensive parts list of functional elements in the human genome, including elements that act at the protein and RNA levels, and regulatory elements that control cells and circumstances in which a gene is active.[17] Data analysis during the pilot phase (2003 - 2007) was coordinated by the Ensembl group, a joint project of EBI and the Wellcome Trust Sanger Institute. During the initial pilot and technology development phases of the project, 44 regions—approximately 1% of the human genome—were targeted for analysis using a variety of experimental and computational methods.[18] All data produced by ENCODE investigators and the results of ENCODE analysis projects from 2003 to 2012 are hosted in the UCSC Genome browser and database. ENCODE results from 2013 and later are freely available for download and analysis from the ENCODE Project Portal. To annotate all evidence-based gene features (genes, transcripts, coding sequences, etc.) in the entire human genome at a high accuracy, ENCODE consortium create the subproject GENCODE.
8.2. Human Genome Project
The Human Genome Project was an international research effort to determine the sequence of the human genome and identify the genes that it contains. The Project was coordinated by the National Institutes of Health and the U.S. Department of Energy. Additional contributors included universities across the United States and international partners in the United Kingdom, France, Germany, Japan, and China. The Human Genome Project formally began in 1990 and was completed in 2003, 2 years ahead of its original schedule.[19] Following the release of the completed human genome sequence in April 2003, the scientific community intensified its efforts to mine the data for clues about how the body works in health and in disease. A basic requirement for this understanding of human biology is the ability to identify and characterize sequence-based functional elements through experimentation and computational analysis. In September 2003, the NHGRI introduced the ENCODE project to facilitate the identification and analysis of the complete set of functional elements in the human genome sequence.[18]
9. Sub Projects
9.1. Ensembl
Ensembl is part of the GENCODE project, and it has played a critical role to provide automatic annotation on the human reference genome assembly and to merge this annotation with manual annotation from the HAVANA team. The gene set provided by Ensembl for human is the GENCODE gene set [20]
9.2. lncRNA Expression Microarray Design
A key research area of the GENCODE project was to investigate the biological significance of long non-coding RNAs (lncRNA). To better understand the lncRNA expression in Humans, a sub project was created by GENCODE to develop custom microarray platforms capable of quantifying the transcripts in the GENCODE lncRNA annotation.[21] A number of designs have been created using the Agilent Technologies eArray system, and these designs are available in a standard custom Agilent format.[22]
9.3. RGASP
The RNA-seq Genome Annotation Assessment Project (RGASP) project is designed to assess the effectiveness of various computational methods for high quality RNA-sequence data analysis. The primary goals of RGASP are to provide an unbiased evaluation for RNA-seq alignment, transcript characterisation (discovery, reconstruction and quantification) software, and to determine the feasibility of automated genome annotations based on transcriptome sequencing.[23]
RGASP is organised in a consortium framework modelled after the EGASP (ENCODE Genome Annotation Assessment Project) gene prediction workshop, and two rounds of workshops have been conducted to address different aspects of RNA-seq analysis as well as changing sequencing technologies and formats. One of the main discoveries from rounds 1 & 2 of the project was the importance of read alignment on the quality of gene predictions produced. Hence, a third round of RGASP workshop is currently being conducted (in 2014) to focus primarily on read mapping to the genome.[24]
References
- "GENCODE – Goals". Wellcome Trust Sanger Institute.. c. 2013. http://www.gencodegenes.org/gencode_goals.html. Retrieved 5 September 2014.
- "GENCODE – Data". Wellcome Trust Sanger Institute.. September 2019. http://www.gencodegenes.org/human/releases.html. Retrieved 14 October 2019.
- The ENCODE Project Consortium (22 October 2004). "The ENCODE (ENCyclopedia of DNA Elements) Project". Science 306 (5696): 636–640. doi:10.1126/science.1105136. PMID 15499007. https://dx.doi.org/10.1126%2Fscience.1105136
- "GENCODE: producing a reference annotation for ENCODE.". Genome Biol 7 Suppl 1: S4.1–9. 2006. doi:10.1186/gb-2006-7-s1-s4. PMID 16925838. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1810553
- The ENCODE Project Consortium (14 June 2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project.". Nature 447 (7146): 799–816. doi:10.1038/nature05874. PMID 17571346. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2212820
- "Researchers Expand Efforts to Explore Functional Landscape of the Human Genome". Wellcome Trust Sanger Institute.. 9 October 2007. http://www.sanger.ac.uk/about/press/2007/071009.html. Retrieved 8 September 2014.
- "GENCODE: the reference human genome annotation for The ENCODE Project.". Genome Res 22 (9): 1760–74. 2012. doi:10.1101/gr.135350.111. PMID 22955987. PMC 3431492. http://repositori.upf.edu/bitstream/10230/22636/1/guigo_genomres4.pdf.
- "GENCODE – Home page". Wellcome Trust Sanger Institute.. c. 2013. http://www.gencodegenes.org/. Retrieved 8 September 2014.
- "Participants, all funded personnel". Wellcome Trust Sanger Institute. c. 2014. http://www.gencodegenes.org/all_participants.html. Retrieved 8 September 2014.
- "GENCODE Project Participants". Genome BioInformatics Research Lab. c. 2005. http://genome.crg.es/gencode/participants.html. Retrieved 8 September 2014.
- "GENCODE – Statistics". Wellcome Trust Sanger Institute. c. 2014. http://www.gencodegenes.org/stats.html. Retrieved 8 September 2014.
- Searle, S et al. (2010). "The GENCODE human gene set.". Genome Biology 11 (Suppl 1): 36. doi:10.1186/gb-2010-11-S1-P36. PMC 3026266. http://genomebiology.com/2010/11/S1/P36.
- "Ensembl Gene Set". August 2014. http://Aug2014.archive.ensembl.org/info/genome/genebuild/genome_annotation.html. Retrieved 6 September 2014.
- Kokocinski, F; Harrow, J; Hubbard, T (2010). "AnnoTrack - a tracking system for genome annotation.". BMC Genomics 11: 538. doi:10.1186/1471-2164-11-538. PMID 20923551. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3091687
- "A system for annotating alternative splice isoforms". July 2014. http://appris.bioinfo.cnio.es/. Retrieved 6 September 2014.
- "What is a gene, post-ENCODE? History and updated definition.". Genome Res 17 (6): 669–81. 2007. doi:10.1101/gr.6339607. PMID 17567988. https://dx.doi.org/10.1101%2Fgr.6339607
- "ENCODE: Encyclopedia of DNA Elements". c. 2014. https://www.encodeproject.org/. Retrieved 7 September 2014.
- "ENCODE: Pilot Project at UCSC". c. 2007. http://genome.ucsc.edu/ENCODE/pilot.html. Retrieved 7 September 2014.
- "The Human Genome Project". U.S. National Library of Medicine (NLM). 1 September 2014. http://ghr.nlm.nih.gov/handbook/hgp/description. Retrieved 7 September 2014.
- "ENCODE data in Ensembl". August 2014. http://www.ensembl.org/info/website/tutorials/encode.html. Retrieved 7 September 2014.
- "The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression.". Genome Res 22 (9): 1775–89. 2012. doi:10.1101/gr.132159.111. PMID 22955988. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3431493
- "GENCODE - lncRNA microarray". c. 2013. http://www.gencodegenes.org/lncrna_microarray.html. Retrieved 10 September 2014.
- "GENCODE - RGASP 1/2 Guidelines". c. 2013. http://www.gencodegenes.org/rgasp/. Retrieved 10 September 2014.
- "GENCODE - RGASP 1/2 Guidelines". c. 2013. http://www.gencodegenes.org/rgasp/rgasp3.html. Retrieved 10 September 2014.


