 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Beatrix Zheng | -- | 2601 | 2022-11-03 01:38:39 |
Video Upload Options
Site-specific recombinase technologies are genome engineering tools that depend on recombinase enzymes to replace targeted sections of DNA.
1. History
In the late 1980s gene targeting in murine embryonic stem cells (ESCs) enabled the transmission of mutations into the mouse germ line, and emerged as a novel option to study the genetic basis of regulatory networks as they exist in the genome. Still, classical gene targeting proved to be limited in several ways as gene functions became irreversibly destroyed by the marker gene that had to be introduced for selecting recombinant ESCs. These early steps led to animals in which the mutation was present in all cells of the body from the beginning leading to complex phenotypes and/or early lethality. There was a clear need for methods to restrict these mutations to specific points in development and specific cell types. This dream became reality when groups in the USA were able to introduce bacteriophage and yeast-derived site-specific recombination (SSR-) systems into mammalian cells as well as into the mouse.[1][2][3]
2. Classification, Properties and Dedicated Applications
Common genetic engineering strategies require a permanent modification of the target genome. To this end great sophistication has to be invested in the design of routes applied for the delivery of transgenes. Although for biotechnological purposes random integration is still common, it may result in unpredictable gene expression due to variable transgene copy numbers, lack of control about integration sites and associated mutations. The molecular requirements in the stem cell field are much more stringent. Here, homologous recombination (HR) can, in principle, provide specificity to the integration process, but for eukaryotes it is compromised by an extremely low efficiency. Although meganucleases, zinc-finger- and transcription activator-like effector nucleases (ZFNs and TALENs) are actual tools supporting HR, it was the availability of site-specific recombinases (SSRs) which triggered the rational construction of cell lines with predictable properties. Nowadays both technologies, HR and SSR can be combined in highly efficient "tag-and-exchange technologies".[4]
Many site-specific recombination systems have been identified to perform these DNA rearrangements for a variety of purposes, but nearly all of these belong to either of two families, tyrosine recombinases (YR) and serine recombinases (SR), depending on their mechanism. These two families can mediate up to three types of DNA rearrangements (integration, excision/resolution, and inversion) along different reaction routes based on their origin and architecture.[5]
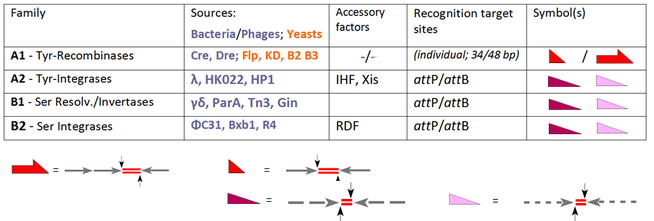
The founding member of the YR family is the lambda integrase, encoded by bacteriophage λ, enabling the integration of phage DNA into the bacterial genome. A common feature of this class is a conserved tyrosine nucleophile attacking the scissile DNA-phosphate to form a 3'-phosphotyrosine linkage. Early members of the SR family are closely related resolvase / DNA invertases from the bacterial transposons Tn3 and γδ, which rely on a catalytic serine responsible for attacking the scissile phosphate to form a 5'-phosphoserine linkage. These undisputed facts, however, were compromised by a good deal of confusion at the time other members entered the scene, for instance the YR recombinases Cre and Flp (capable of integration, excision/resolution as well as inversion), which were nevertheless welcomed as new members of the "integrase family". The converse examples are PhiC31 and related SRs, which were originally introduced as resolvase/invertases although, in the absence of auxiliary factors, integration is their only function. Nowadays the standard activity of each enzyme determines its classification reserving the general term "recombinase" for family members which, per se, comprise all three routes, INT, RES and INV:
Our table extends the selection of the conventional SSR systems and groups these according to their performance. All of these enzymes recombine two target sites, which are either identical (subfamily A1) or distinct (phage-derived enzymes in A2, B1 and B2).[6] Whereas for A1 these sites have individual designations ("FRT" in case of Flp-recombinase, loxP for Cre-recombinase), the terms "attP" and "attB" (attachment sites on the phage and bacterial part, respectively) are valid in the other cases. In case of subfamily A1 we have to deal with short (usually 34 bp-) sites consisting of two (near-)identical 13 bp arms (arrows) flanking an 8 bp spacer (the crossover region, indicated by red line doublets).[7] Note that for Flp there is an alternative, 48 bp site available with three arms, each accommodating a Flp unit (a so-called "protomer"). attP- and attB-sites follow similar architectural rules, but here the arms show only partial identity (indicated by the broken lines) and differ in both cases. These features account for relevant differences:
- recombination of two identical educt sites leads to product sites with the same composition, although they contain arms from both substrates; these conversions are reversible;
- in case of attP x attB recombination crossovers can only occur between these complementary partners in processes that lead to two different products (attP x attB → attR + attL) in an irreversible fashion.
In order to streamline this chapter the following implementations will be focused on two recombinases (Flp and Cre) and just one integrase (PhiC31) since their spectrum covers the tools which, at present, are mostly used for directed genome modifications. This will be done in the framework of the following overview.
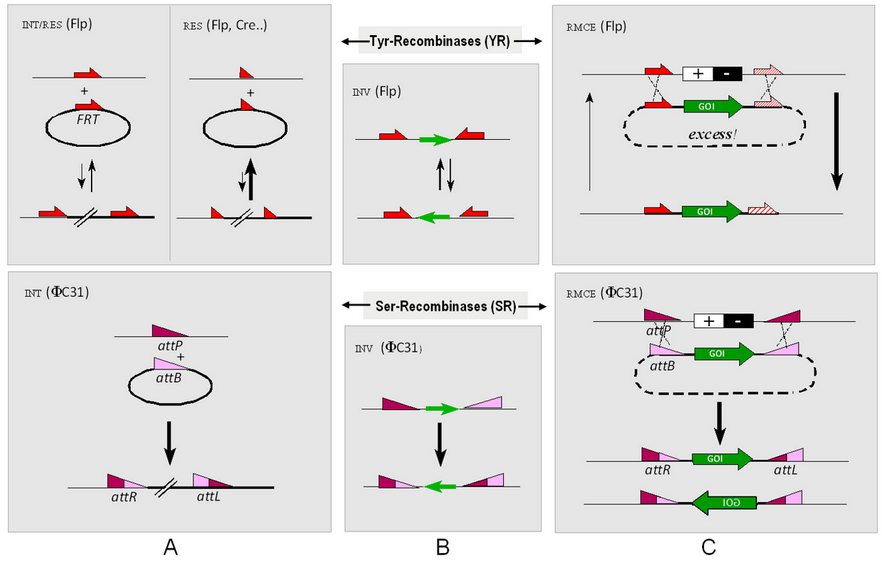
3. Reaction Routes
The mode integration/resolution and inversion (INT/RES and INV) depend on the orientation of recombinase target sites (RTS), among these pairs of attP and attB. Section C indicates, in a streamlined fashion, the way recombinase-mediated cassette exchange (RMCE) can be reached by synchronous double-reciprocal crossovers (rather than integration, followed by resolution).[8][9]
Tyr-Recombinases are reversible, while the Ser-Integrase is unidirectional. Of note is the way reversible Flp (a Tyr recombinase) integration/resolution is modulated by 48 bp (in place of 34 bp minimal) FRT versions: the extra 13 bp arm serves as a Flp "landing path" contributing to the formation of the synaptic complex, both in the context of Flp-INT and Flp-RMCE functions (see the respective equilibrium situations). While it is barely possible to prevent the (entropy-driven) reversion of integration in section A for Cre and hard to achieve for Flp, RMCE can be completed if the donor plasmid is provided at an excess due to the bimolecular character of both the forward- and the reverse reaction. Posing both FRT sites in an inverse manner will lead to an equilibrium of both orientations for the insert (green arrow). In contrast to Flp, the Ser integrase PhiC31 (bottom representations) leads to unidirectional integration, at least in the absence of an recombinase-directionality (RDF-)factor.[10] Relative to Flp-RMCE, which requires two different ("heterospecific") FRT-spacer mutants, the reaction partner (attB) of the first reacting attP site is hit arbitrarily, such that there is no control over the direction the donor cassette enters the target (cf. the alternative products). Also different from Flp-RMCE, several distinct RMCE targets cannot be mounted in parallel, owing to the lack of heterospecific (non-crossinteracting) attP/attB combinations.
3.1. Cre Recombinase
Cre recombinase (Cre) is able to recombine specific sequences of DNA without the need for cofactors. The enzyme recognizes 34 base pair DNA sequences called loxP ("locus of crossover in phage P1"). Depending on the orientation of target sites with respect to one another, Cre will integrate/excise or invert DNA sequences. Upon the excision (called "resolution" in case of a circular substrate) of a particular DNA region, normal gene expression is considerably compromised or terminated.[11]
Due to the pronounced resolution activity of Cre, one of its initial applications was the excision of loxP-flanked ("floxed") genes leading to cell-specific gene knockout of such a floxed gene after Cre becomes expressed in the tissue of interest. Current technologies incorporate methods, which allow for both the spatial and temporal control of Cre activity. A common method facilitating the spatial control of genetic alteration involves the selection of a tissue-specific promoter to drive Cre expression. Placement of Cre under control of such a promoter results in localized, tissue-specific expression. As an example, Leone et al. have placed the transcription unit under the control of the regulatory sequences of the myelin proteolipid protein (PLP) gene, leading to induced removal of targeted gene sequences in oligodendrocytes and Schwann cells.[12] The specific DNA fragment recognized by Cre remains intact in cells, which do not express the PLP gene; this in turn facilitates empirical observation of the localized effects of genome alterations in the myelin sheath that surround nerve fibers in the central nervous system (CNS) and the peripheral nervous system (PNS).[13] Selective Cre expression has been achieved in many other cell types and tissues as well.
In order to control temporal activity of the excision reaction, forms of Cre which take advantage of various ligand binding domains have been developed. One successful strategy for inducing specific temporal Cre activity involves fusing the enzyme with a mutated ligand-binding domain for the human estrogen receptor (ERt). Upon the introduction of tamoxifen (an estrogen receptor antagonist), the Cre-ERt construct is able to penetrate the nucleus and induce targeted mutation. ERt binds tamoxifen with greater affinity than endogenous estrogens, which allows Cre-ERt to remain cytoplasmic in animals untreated with tamoxifen. The temporal control of SSR activity by tamoxifen permits genetic changes to be induced later in embryogenesis and/or in adult tissues.[12] This allows researchers to bypass embryonic lethality while still investigating the function of targeted genes.
Recent extensions of these general concepts led to generating the "Cre-zoo", i.e. collections of hundreds of mouse strains for which defined genes can be deleted by targeted Cre expression.[3]
3.2. Flp Recombinase
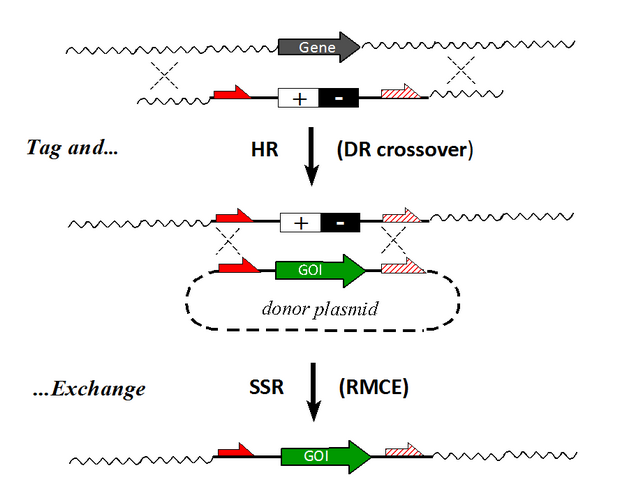
In its natural host (S. cerevisiae) the Flp/FRT system enables replication of a "2μ plasmid" by the inversion of a segment that is flanked by two identical, but oppositely oriented FRT sites ("flippase" activity). This inversion changes the relative orientation of replication forks within the plasmid enabling "rolling circle"—amplification of the circular 2μ entity before the multimeric intermediates are resolved to release multiple monomeric products. Whereas 34 bp minimal FRT sites favor excision/resolution to a similar extent as the analogue loxP sites for Cre, the natural, more extended 48 bp FRT variants enable a higher degree of integration, while overcoming certain promiscuous interactions as described for phage enzymes like Cre- [5] and PhiC31.[6] An additional advantage is the fact, that simple rules can be applied to generate heterospecific FRT sites which undergo crossovers with equal partners but nor with wild type FRTs. These facts have enabled, since 1994, the development and continuous refinements of recombinase-mediated cassette exchange (RMCE-)strategies permitting the clean exchange of a target cassette for an incoming donor cassette.[6]
Based on the RMCE technology, a particular resource of pre-characterized ES-strains that lends itself to further elaboration has evolved in the framework of the EUCOMM (European Conditional Mouse Mutagenesis) program, based on the now established Cre- and/or Flp-based "FlExing" (Flp-mediated excision/inversion) setups,[6] involving the excision and inversion activities. Initiated in 2005, this project focused first on saturation mutagenesis to enable complete functional annotation of the mouse genome (coordinated by the International Knockout-Mouse Consortium, IKMC) with the ultimate goal to have all protein genes mutated via gene trapping and -targeting in murine ES cells.[14] These efforts mark the top of various "tag-and-exchange" strategies, which are dedicated to tagging a distinct genomic site such that the "tag" can serve as an address to introduce novel (or alter existing) genetic information. The tagging step per se may address certain classes of integration sites by exploiting integration preferences of retroviruses or even site specific integrases like PhiC31, both of which act in an essentially unidirectional fashion.
The traditional, laborious "tag-and-exchange" procedures relied on two successive homologous recombination (HR-)steps, the first one ("HR1") to introduce a tag consisting of a selection marker gene. "HR2" was then used to replace the marker by the "GOI. In the first ("knock-out"-) reaction the gene was tagged with a selectable marker, typically by insertion of a hygtk ([+/-]) cassette providing G418 resistance. In the following "knock-in" step, the tagged genomic sequence was replaced by homologous genomic sequences with certain mutations. Cell clones could then be isolated by their resistance to ganciclovir due to loss of the HSV-tk gene, i.e. ("negative selection"). This conventional two-step tag-and-exchange procedure [15] could be streamlined after the advent of RMCE, which could take over and add efficiency to the knock-in step.
3.3. PhiC31 Integrase
Without much doubt, Ser integrases are the current tools of choice for integrating transgenes into a restricted number of well-understood genomic acceptor sites that mostly (but not always) mimic the phage attP site in that they attract an attB-containing donor vector. At this time the most prominent member is PhiC31-INT with proven potential in the context of human and mouse genomes.
Contrary to the above Tyr recombinases, PhiC31-INT as such acts in a unidirectional manner, firmly locking in the donor vector at a genomically anchored target. An obvious advantage of this system is that it can rely on unmodified, native attP (acceptor) and attB donor sites. Additional benefits (together with certain complications) may arise from the fact that mouse and human genomes per se contain a limited number of endogenous targets (so called "attP-pseudosites"). Available information suggests that considerable DNA sequence requirements let the integrase recognize fewer sites than retroviral or even transposase-based integration systems opening its career as a superior carrier vehicle for the transport and insertion at a number of well established genomic sites, some of which with so called "safe-harbor" properties.[10]
Exploiting the fact of specific (attP x attB) recombination routes, RMCE becomes possible without requirements for synthetic, heterospecific att-sites. This obvious advantage, however comes at the expense of certain shortcomings, such as lack of control about the kind or directionality of the entering (donor-) cassette.[6] Further restrictions are imposed by the fact that irreversibility does not permit standard multiplexing-RMCE setups including "serial RMCE" reactions, i.e., repeated cassette exchanges at a given genomic locus.
4. Outlook and Perspectives
Annotation of the human and mouse genomes has led to the identification of >20 000 protein-coding genes and >3 000 noncoding RNA genes, which guide the development of the organism from fertilization through embryogenesis to adult life. Although dramatic progress is noted, the relevance of rare gene variants has remained a central topic of research.
As one of the most important platforms for dealing with vertebrate gene functions on a large scale, genome-wide genetic resources of mutant murine ES cells have been established. To this end four international programs aimed at saturation mutagenesis of the mouse genome have been founded in Europe and North America (EUCOMM, KOMP, NorCOMM, and TIGM). Coordinated by the International Knockout Mouse Consortium (IKSC) these ES-cell repositories are available for exchange between international research units. Present resources comprise mutations in 11 539 unique genes, 4 414 of these conditional.[14]
The relevant technologies have now reached a level permitting their extension to other mammalian species and to human stem cells, most prominently those with an iPS (induced pluripotent) status.
References
- Sauer, Brian; Henderson, Nancy (1988). "Site-Specific DNA Recombination in Mammalian Cells by the Cre Recombinase of Bacteriophage P1". Proceedings of the National Academy of Sciences of the United States of America 85 (14): 5166–70. doi:10.1073/pnas.85.14.5166. PMID 2839833. Bibcode: 1988PNAS...85.5166S. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=281709
- O'Gorman, Stephen; Fox, Daniel T.; Wahl, Geoffrey M. (1991). "Recombinase-mediated gene activation and site-specific integration in mammalian cells". Science 251 (4999): 1351–5. doi:10.1126/science.1900642. PMID 1900642. Bibcode: 1991Sci...251.1351O. https://dx.doi.org/10.1126%2Fscience.1900642
- Rajewsky, Klaus (2007). "From a Dream to Reality". European Journal of Immunology 37: S134–7. doi:10.1002/eji.200737819. PMID 17972357. https://dx.doi.org/10.1002%2Feji.200737819
- Branda, Catherine S.; Dymecki, Susan M. (2004). "Talking about a RevolutionThe Impact of Site-Specific Recombinases on Genetic Analyses in Mice". Developmental Cell 6 (1): 7–28. doi:10.1016/S1534-5807(03)00399-X. PMID 14723844. https://dx.doi.org/10.1016%2FS1534-5807%2803%2900399-X
- Nern, A.; Pfeiffer, B. D.; Svoboda, K.; Rubin, G. M. (2011). "Multiple new site-specific recombinases for use in manipulating animal genomes". Proceedings of the National Academy of Sciences 108 (34): 14198–203. doi:10.1073/pnas.1111704108. PMID 21831835. Bibcode: 2011PNAS..10814198N. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3161616
- Turan, S.; Bode, J. (2011). "Site-specific recombinases: From tag-and-target- to tag-and-exchange-based genomic modifications". The FASEB Journal 25 (12): 4088–107. doi:10.1096/fj.11-186940. PMID 21891781. https://dx.doi.org/10.1096%2Ffj.11-186940
- Bode, Jürgen; Schlake, Thomas; Iber, Michaela; Schübeler, Dirk; Seibler, Jost; Snezhkov, Evgeney; Nikolaev, Lev (2000). "The Transgeneticists Toolbox: Novel Methods for the Targeted Modification of Eukaryotic Genomes". Biological Chemistry 381 (9–10): 801–13. doi:10.1515/BC.2000.103. PMID 11076013. https://dx.doi.org/10.1515%2FBC.2000.103
- Lyznik, Leszek A.; Mitchell, Jon C.; Hirayama, Lynne; Hodges, Thomas K. (1993). "Activity of yeast FLP recombinase in maize and rice protoplasts". Nucleic Acids Research 21 (4): 969–75. doi:10.1093/nar/21.4.969. PMID 8451196. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=309231
- Lauth, M.; Spreafico, F; Dethleffsen, K; Meyer, M (2002). "Stable and efficient cassette exchange under non-selectable conditions by combined use of two site-specific recombinases". Nucleic Acids Research 30 (21): 115e. doi:10.1093/nar/gnf114. PMID 12409474. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=135837
- Karow, Marisa; Calos, Michele P (2011). "The therapeutic potential of phiC31 integrase as a gene therapy system". Expert Opinion on Biological Therapy 11 (10): 1287–96. doi:10.1517/14712598.2011.601293. PMID 21736536. https://dx.doi.org/10.1517%2F14712598.2011.601293
- Oumard, André; Qiao, Junhua; Jostock, Thomas; Li, Jiandong; Bode, Juergen (2006). "Recommended Method for Chromosome Exploitation: RMCE-based Cassette-exchange Systems in Animal Cell Biotechnology". Cytotechnology 50 (1–3): 93–108. doi:10.1007/s10616-006-6550-0. PMID 19003073. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3476001
- Leone, Dino P; Genoud, S.Téphane; Atanasoski, Suzana; Grausenburger, Reinhard; Berger, Philipp; Metzger, Daniel; MacKlin, Wendy B; Chambon, Pierre et al. (2003). "Tamoxifen-inducible glia-specific Cre mice for somatic mutagenesis in oligodendrocytes and Schwann cells". Molecular and Cellular Neuroscience 22 (4): 430–40. doi:10.1016/S1044-7431(03)00029-0. PMID 12727441. https://dx.doi.org/10.1016%2FS1044-7431%2803%2900029-0
- Koenning, M.; Jackson, S.; Hay, C. M.; Faux, C.; Kilpatrick, T. J.; Willingham, M.; Emery, B. (2012). "Myelin Gene Regulatory Factor is Required for Maintenance of Myelin and Mature Oligodendrocyte Identity in the Adult CNS". Journal of Neuroscience 32 (36): 12528–42. doi:10.1523/JNEUROSCI.1069-12.2012. PMID 22956843. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3752083
- Bradley, Allan; Anastassiadis, Konstantinos; Ayadi, Abdelkader; Battey, James F.; Bell, Cindy; Birling, Marie-Christine; Bottomley, Joanna; Brown, Steve D. et al. (2012). "The mammalian gene function resource: The international knockout mouse consortium". Mammalian Genome 23 (9–10): 580–6. doi:10.1007/s00335-012-9422-2. PMID 22968824. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3463800
- Askew, G. Roger; Doetschman, Thomas; Lingrel, Jerry B. (1993). "Site-directed point mutations in embryonic stem cells: A gene-targeting tag-and-exchange strategy". Molecular and Cellular Biology 13 (7): 4115–24. doi:10.1128/MCB.13.7.4115. PMID 8391633. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=359961

