 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alice Vassiliou | + 4895 word(s) | 4895 | 2020-11-27 07:59:15 | | | |
| 2 | Rita Xu | -1922 word(s) | 2973 | 2020-11-30 04:52:00 | | |
Video Upload Options
The pulmonary endothelium is a metabolically active continuous monolayer of squamous endothelial cells that internally lines blood vessels and mediates key processes involved in lung homoeostasis. Many of these processes are disrupted in acute respiratory distress syndrome (ARDS), which is marked among others by diffuse endothelial injury, intense activation of the coagulation system and increased capillary permeability. Most commonly occurring in the setting of sepsis, ARDS is a devastating illness, associated with increased morbidity and mortality and no effective pharmacological treatment. Endothelial cell damage has an important role in the pathogenesis of ARDS and several biomarkers of endothelial damage have been tested in determining prognosis. By further understanding the endothelial pathobiology, development of endothelial-specific therapeutics might arise. In this review, we will discuss the underlying pathology of endothelial dysfunction leading to ARDS and emerging therapies. Furthermore, we will present a brief overview demonstrating that endotheliopathy is an important feature of hospitalised patients with coronavirus disease-19 (COVID-19).
1. Introduction
Using the updated Berlin definition, acute respiratory distress syndrome (ARDS) is defined as a syndrome of acute onset, with bilateral diffuse infiltrates on chest radiography, and non-cardiogenic respiratory failure, leading to mild, moderate, or severe oxygenation impairment [1]. Pathophysiologically, it is characterized by damage to the capillary endothelium and alveolar epithelium, and fluid accumulation in the alveolar space, leading to alveolar oedema. These changes in the microvascular endothelial structure and function play a central role in the acute inflammatory response, in which the body tries to eliminate microbial invaders. To achieve this, the endothelium becomes leaky and inflamed, allowing innate immune cells and humoral effector molecules to cross the barrier to the site of infection [2]. When the phenomenon becomes overwhelming, it leads to ARDS, whose inciting events can be either direct (mainly pneumonia, aspiration of gastric contents) or indirect (mainly sepsis, multiple trauma) insults to the lung, with sepsis and pneumonia being the most common cause of ARDS in humans [3]. The incidence of ARDS varies widely, from 15 to 70 cases per 100,000 persons per year, representing approximately 5% of hospitalized, mechanically ventilated patients [4]. Understanding the molecular mechanisms of endothelial dysfunction has potential diagnostic, prognostic, and therapeutic implications for this fatal disease [5][6][7]. The old term acute lung injury (ALI) was used in clinical studies, along with the term ARDS, until the Berlin definition was released, and it is still being used in experimental models [3].
2. Pathogenesis
A single layer of endothelial cells (ECs) lines the entire vascular system, and this vascular endothelium forms the innermost layer of all blood vessels. In the past, the vascular endothelium was considered to be inert and nothing more than a nucleated layer. However, it is now clear that it actively participates in several key functions including angiogenesis, blood clotting, vasomotor tone, and inflammation [8]. Moreover, endothelial cells produce various cytokines and adhesion molecules [9].
As discussed in detail below, endothelial dysfunction is characterized by a change in EC functions. These include increased permeability leading to vascular leakage and oedema formation; disruption of the balance between vasodilators and vasoconstrictors; pro-inflammatory characteristics, including increased expression of adhesion molecules, receptors and signal transduction molecules, as well as release of reactive oxygen species; pro-coagulant and anti-fibrinolytic phenotype, miscommunication with adjacent vascular cell wall.
3. Pulmonary Endothelial Functions
The vascular endothelium is a highly specialized metabolically active organ with many physiological, immunological, and synthesizing functions (Table 1). In this review, we will focus on changes occurring in some of these functions in ARDS (Figure 1). We will discuss these changes in both the clinical and preclinical context. Most animal models of ALI are based on clinical disorders that can lead to the development of ARDS in humans, such as sepsis, trauma, aspiration of gastric contents, and reperfusion of ischemic tissues. The animal models reproducing these risk factors and most suitable for the study of ARDS are related to ventilator-induced lung injury (VILI), lipopolysaccharide (LPS), live bacteria, hyperoxia, bleomycin (BLM), oleic acid, cecal ligation and puncture and acid aspiration [10].
Table 1. Major pulmonary endothelial functions.
|
Barrier and transport functions |
|
Synthesis of vasoactive compounds–maintenance of vascular tone |
|
Host defence—production of cytokines and chemokines |
|
Haemostasis and coagulation |
|
Angiogenesis—production of growth factors |
|
Expression of receptors and signal transduction molecules |
|
Expression of adhesion molecules |
|
Production of reactive oxygen species |
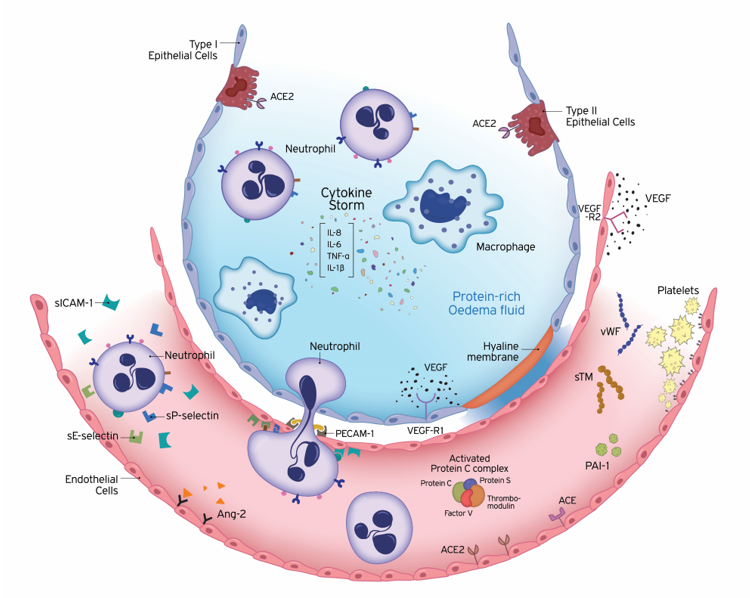
Figure 1. Underlying pathology of endothelial dysfunction leading to acute respiratory distress syndrome (ARDS). ARDS is characterized by damage to the capillary endothelium and alveolar epithelium. Disruption of the endothelial barrier results in the movement of fluid and macromolecules into the interstitial space and pulmonary air spaces causing pulmonary oedema. The formation of a hyaline membrane in alveolar walls allows exudation of neutrophils and protein-rich fluid into the alveolar space. Transport across the endothelium can occur either via the endothelial cell (transcellular) or between adjacent cells, through inter-endothelial junctions (IEJs) (paracellular). The changes in the microvascular endothelial structure and function play a central role in the acute inflammatory response, in which the body tries to eliminate microbial invaders. To achieve this, the endothelium becomes leaky and inflamed, allowing innate immune cells and humoral effector molecules to cross the barrier to the site of infection. This defence mechanism may become deleterious, under overwhelming pathological conditions, leading to ARDS. The cells of the innate immune system release large amounts of pro- and anti-inflammatory cytokines, such as IL-1β, IL-6, IL-8, and TNF-α. The high levels of circulating cytokines can potentiate organ damage by endothelial injury and other routes. Endothelial damage is associated with activation of neutrophils and expression of neutrophil and endothelial adhesion molecules. E-selectin and P-selectin are early mediators of the adhesion of activated neutrophils to endothelia in inflammatory states, prior to their firm adhesion and diapedesis at sites of tissue injury and inflammation. Intercellular adhesion molecule-1 (ICAM-1) controls the firm adhesion of neutrophils on the endothelium and facilitates their subsequent transendothelial migration via the platelet-endothelial cell adhesion molecule-1 (PECAM-1) to infection sites. In addition to inflammation, coagulation and fibrinolysis are also critical host responses to infection and injury involved in ARDS. Endothelial cells (ECs) coordinate this response by shifting from their normal anti-thrombotic, anti-inflammatory, and pro-fibrinolytic phenotype to an activated state of endothelial dysfunction. ECs actively regulate haemostasis by producing a variety of proteins, including pro-thrombotic substances (von Willebrand factor, P-selectin), molecules restricting coagulation (thrombomodulin) and fibrinolytic factors (plasminogen activators). Plasminogen activator inhibitor-1 (PAI-1) is the major inhibitor of fibrinolysis, whose upregulation leads to a shift from pro- to anti-fibrinolytic phenotypes. The protein C (PC) system provides important control of coagulation by virtue of the capacity of activated protein C (APC) to proteolytically inactivate the cofactors Va and VIIIa. The PC anticoagulant system also involves protein S, and the endothelial receptor thrombomodulin (TM). Conversion of protein C to the anti-coagulant APC is generated by TM-bound thrombin. The vascular endothelium has an important metabolic function with respect to vasoactive substances. Several vasoconstrictors and vasodilators are produced by the endothelium, such as endothelin-1, angiotensin-2, nitric oxide, and prostacyclin, which regulate vasomotor tone and the recruitment and activity of inflammatory cells and regulate thrombosis. In addition to breaking down bradykinin, ACE hydrolyses angiotensin I to angiotensin II and the balance between ACE and ACE2 has been suggested to be crucial for controlling angiotensin II levels. Vascular development strongly depends on the collaboration of growth factors. Vascular endothelial growth factor (VEGF) is a glycoprotein originally isolated as a permeability factor with unique specificity for vascular ECs. Angiopoietin-2 (Ang-2) disrupts the protective effects of Ang-1-Tie2 signalling, promoting vascular leakage. Ang-1, angiopoietin-1; Ang-2, angiopoietin-2; APC, activated protein C; IEJs, inter-endothelial junctions; IL, interleukin; PAI-1, plasminogen activator inhibitor 1; PECAM-1, platelet-endothelial cell adhesion molecule-1; sICAM-1, soluble intercellular adhesion molecule-1; sTM, soluble thrombomodulin; TNF-α, tumour necrosis factor-alpha; VEGF, vascular endothelial growth factor; VEGF-R, VEGF receptor; vWF, von Willebrand factor.
3.1. Endothelium Barrier and Transport Functions
The endothelium, apart from being a semipermeable barrier separating blood from the surrounding tissues and, in the lungs, blood from the air, also regulates the transport of fluid and solutes between the blood and the interstitial space [8]. Disruption of the endothelial barrier results in the movement of fluid and macromolecules into the interstitial space and pulmonary air spaces causing pulmonary oedema. Transport across the endothelium can occur either via the endothelial cell (transcellular) or between adjacent cells, through inter-endothelial junctions (IEJs) (paracellular) [11][12]. IEJs are composed of tight junctions (TJs), adherens junctions (AJs), and gap junctions (GJs), which interact with integrin receptors to support EC adhesion to the underlying matrix [13]. TJs, formed by occludin, claudins, and junctional adhesion molecules (JAMs), act as a selective barrier to the entrance of molecules from the circulation; AJs, formed by vascular endothelial cadherin (VE-cadherin), mediate cell-to-cell contact and have a central role in barrier function, while GJs, which are formed by connexins, facilitate direct cell-to-cell transfer of signalling molecules, ions, and transmembrane potential [13][14]. Solutes and water can also cross the endothelial barrier via a transcellular pathway. Vesicles (or caveolae) have long been considered a pathway for the exchange of plasma proteins between the blood and interstitial compartment [15]. Although vascular permeability depends on both the tight junctions and caveolae, oedema develops mainly as a result of dysfunction of tight junctions [9]. Persistent opening of intercellular junctions leads to the formation of protein-rich oedema in the interstitial tissue, the main characteristic of tissue inflammation that may cause fatal diseases such as ARDS [16]. Thus, understanding the signalling pathways that prevent the disruption of endothelial barrier functions will be important for reversing ARDS and other diseases occurring as a consequence of such disruption. Indeed, a very recent study has shown that treatment with unfractionated heparin alleviated sepsis-induced lung injury in vivo by protecting TJs in lung microvascular endothelial cells (LMVECs) [17]. IEJs are also covered by a layer of fibrous matrix, the endothelial glycocalyx (EG). Dysfunction of the glycocalyx can also cause microvascular leakage, and evidence of EG shedding was discovered in ARDS established after flu syndrome [18]. Moreover, the carotenoid chemical compound, crocin, alleviated LPS-induced ARDS by means of protecting against glycocalyx damage [19].
3.2. Vascular Tone
The vascular endothelium has an important metabolic function with respect to vasoactive substances. Several vasoconstrictors and vasodilators are produced by the endothelium, such as endothelin-1, angiotensin-2, nitric oxide, and prostacyclin, which regulate vasomotor tone and the recruitment and activity of inflammatory cells and regulate thrombosis [20]. The normal balance between pulmonary vasodilators and vasoconstrictors is disrupted in ALI in favour of the latter, and thus results in increased pulmonary vascular resistance and pulmonary hypertension [21].
3.2.1. Endothelin-1
Endothelin-1 (ET-1), a potent vasoconstrictor peptide produced by endothelial cells and degraded predominantly in the pulmonary vasculature, has long been implicated in the development of lung injury. ET-1 concentrations are elevated in ARDS as the result of both increased formation and decreased disposal [22]. In critically ill patients with sepsis, including ARDS subjects, increased endothelin production may contribute to local increases in vascular resistance, hypoperfusion, and the development of organ failure [23]. Patients with ARDS have increased plasma endothelin-1 levels, associated with abnormal pulmonary endothelin-1 metabolism. These abnormalities reverse in patients who recover [24]. It has been suggested that raised circulating ET-1 levels may partly contribute to the development of pulmonary vasoconstriction and bronchoconstriction associated with acute respiratory failure [25]. ET-1 has also been found in the lungs of subjects who died with ARDS, interestingly along with a decrease in both endothelial nitric oxide synthase and inducible nitric oxide synthase in the lung [26]. It has also been demonstrated that in patients with ARDS, ET-1 is produced mainly in the lung and is associated not only with pulmonary vasoconstriction but also the development of permeability oedema, leading to the impairment of oxygenation [27].
In experimental models of ARDS, ET-1 could contribute to pulmonary hypertension seen in acute lung injury [28], while the production of both ET-1 and nitric oxide (NO) was increased in serum and lung tissue in a VILI model [29]. ET-1 was released in an experimental model of oleic acid-induced lung injury [30]. ET-1 has been shown to be downregulated at a transcriptional and translational level by angiopoietin-1 (Ang-1) in both in vitro and in vivo systems, and moreover, cell-based Ang-1 gene transfer markedly ameliorated inflammation in vivo in an experimental model of ARDS. It was suggested that cell-based gene transfer of Ang-1 may provide a novel treatment strategy for ARDS by attenuating vascular inflammation via suppression of ET-1 [31].
Endothelin inhibitors and/or endothelin receptor blockade have also provided further evaluation for the involvement of ET-1 in lung injury and the use of ET-1 suppressors as potential treatments for inflammatory lung diseases. More specifically, phosphoramidon, an endothelin-converting enzyme inhibitor, attenuated LPS-induced ALI [32]; non-selective ET-1 receptor blockade by tezosentan attenuated lung injury in endotoxaemic sheep [33] and alpha-naphthylthiourea-induced lung injury in rats [34]; the P1/fl peptide that selectively antagonises endothelin-A receptors attenuated LPS-induced pulmonary NO production [35]; furthermore, the highly selective ET-1 receptor A inhibitor, sitaxentan, prevented BLM-induced pulmonary inflammation and fibrosis in a murine model [36].
3.2.2. Renin–Angiotensin–Aldosterone System (RAAS)
Angiotensin converting enzyme (ACE), the key RAAS enzyme, is highly expressed on the surface of pulmonary microvascular EC [37]. ACE hydrolyses angiotensin I to angiotensin II and breaks down bradykinin, while its analogue, ACE2, converts angiotensin II into angiotensin (1–7). Angiotensin II exerts powerful vasoconstricting, pro-fibrotic, and pro-inflammatory effects, while angiotensin (1–7) is a potent vasodilator, anti-apoptotic, and anti-proliferative agent [38]. Therefore, ACE2 is considered a negative regulator of the classical ACE [39]. Results of both clinical and experimental studies have provided evidence for the implication of RAAS, and in particular of ACE, in the pathogenesis of acute lung injury.
Clinical cohort studies have suggested the possible involvement of ACE in patients with ARDS. Plasma soluble ACE activity is decreased in ARDS patients [40] and the authors speculated that the decreased ACE levels in sepsis-induced ARDS are due to the presence of circulating inhibitors of ACE. Serum ACE levels were decreased and closely correlated with the severity of lung injury [41], while ACE2 activity was reduced in patients succumbing to ARDS [42]. Bronchoalveolar lavage fluid (BALF) ACE was elevated in ARDS patients with infectious causes of lung injury, possibly reflecting endothelial damage or local increase in ACE production in response to sepsis [43]. Thus, it has been suggested that the balance between ACE and ACE2 is crucial for controlling angiotensin II levels. ACE and ACE2 also appear to modify the severity of ARDS, with ACE2 playing a protective role [44]. A recent study has shown that the number of ACE-positive microvascular circulating endothelial microparticles (EMPs) were a prognostic marker for the development of ARDS in septic patients [45].
Various experimental studies have also demonstrated altered ACE activity in lung injury models. Angiotensin II induces pulmonary oedema in rabbits [46], while in a rat model of smoke inhalation-induced ARDS, inflammation pulmonary oedema and histological changes were possibly attributed to abnormal expression of ACE and ACE2 related pathway [47]. Another study has suggested that the reduced pulmonary microvascular endothelial ACE expression observed in septic ARDS is caused by a two-step process, involving an initially increased shedding of ACE followed by a compensatory downregulation of ACE mRNA and protein expression [48]. ACE2 gene deletion worsens bleomycin-induced lung injury, whereas ACE2 protects against BLM-induced fibrosis. Hence, recombinant ACE2 may have therapeutic potential to reduce respiratory morbidity in ALI/ARDS [49]. ARDS is developed, in part, due to reduced pulmonary levels of angiotensin (1–7), and repletion of this peptide or an angiotensin II receptor antagonist can halt the development of ARDS [50]. It has also been reported that ACE2 and the angiotensin II type 2 receptor protect animals from severe acute lung injury induced by acid aspiration or LPS [51][52].
ACE2 was unexpectedly shown to act as the receptor for the severe acute respiratory syndrome (SARS) virus. It is now known that cells with increased expression of ACE2 have a higher probability to be infected by the new SARS coronavirus 2 (SARS-CoV2) also. Upregulation of ACE2 expression and function is increasingly recognized as a potential therapeutic strategy in hypertension and cardiovascular disease, diabetes, lung injury, and fibrotic disorders. Quantitative mRNA expression profiling of ACE2 showed expression in the thyroid and adrenal glands, and the pancreas [53][54].
The human ACE gene (DCP1) contains a restriction fragment length polymorphism within the coding sequence defined by the presence (insertion, I) or absence (deletion, D) of a 287-bp repeat. The human ACE2 D allele confers increased ACE activity [55]. ACE I/D polymorphism has been shown to be associated with predisposition and prognosis in ARDS [56], while another study has shown that in ARDS patients, it acts as an independent risk factor for mortality [57]. A possible association between the ACE I/D polymorphism genotype and the mortality risk of ALI/ARDS in Asians has been also demonstrated [58], whereas in Chinese patients, the ACE I/D polymorphism is a significant prognostic factor for the outcome of ARDS, patients with the II genotype have a significantly better chance of survival; however, patients with the D allele do not have an increased risk for ARDS [59]. Other studies have failed to show any association. In paediatric ARDS, data did not support the association between ACE I/D genotype and ARDS, although severe hypoxemia was less frequent in D allele carriers, and ACE I/D polymorphism modified angiotensin-II levels [60]. Additionally, another study had data that did not support an association of the ACE gene I/D polymorphism with susceptibility or mortality in severe sepsis or with sepsis-induced ARDS in Spanish patients [61]. ACE activity may be the highest in patients with the DD genotype; however, its concentration and activity are also influenced by other mechanisms, in addition to the genotype; this might possibly explain the differing results regarding the ACE I/D polymorphism and ARDS susceptibility in the reports mentioned above.
The use of angiotensin II receptor blockers or ACE inhibitors has been shown to decrease lung injury in various animal models (reviewed in [62]); however, such a treatment in humans could lead to systemic hypotension [63]. Since ACE2 protects the lung from developing ARDS and functions as a coronavirus receptor for SARS [64], the recombinant ACE2 (rACE2) protein may have an important place in protecting ARDS patients and as a potential therapeutic approach in the management of emerging lung diseases [65].
References
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome. JAMA 2012, 307, 2526–2533, doi:10.1001/jama.2012.5669.
- Martin, T.R. Lung Cytokines and ARDS. Chest 1999, 116, 2S–8S, doi:10.1378/chest.116.suppl_1.2s.
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. J. Respir. Crit. Care Med. 1994, 149 Pt 1, 818–824.
- Sweatt, A.J.; Levitt, J.E. Evolving Epidemiology and Definitions of the Acute Respiratory Distress Syndrome and Early Acute Lung Injury. Chest Med. 2014, 35, 609–624, doi:10.1016/j.ccm.2014.08.002.
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Pharmacol. 2008, 49, 119–133, doi:10.1016/j.vph.2008.06.009.
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Opin. Crit. Care 2008, 14, 22–30, doi:10.1097/mcc.0b013e3282f269b9.
- Orfanos, S.E.; Mavrommati, I.; Korovesi, I.; Roussos, C. Pulmonary endothelium in acute lung injury: From basic science to the critically ill. Intensive Care Med. 2004, 30, 1702–1714, doi:10.1007/s00134-004-2370-x.
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Rev. 2006, 86, 279–367, doi:10.1152/physrev.00012.2005.
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. J. Anaesth. 2004, 93, 105–113, doi:10.1093/bja/aeh163.
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. J. Physiol. Cell. Mol. Physiol. 2008, 295, L379–L399, doi:10.1152/ajplung.00010.2008.
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. N. Y. Acad. Sci. 2008, 1123, 134–145.
- Komarova, Y.; Malik, A.B. Regulation of Endothelial Permeability via Paracellular and Transcellular Transport Pathways. Rev. Physiol. 2010, 72, 463–493, doi:10.1146/annurev-physiol-021909-135833.
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms Regulating Endothelial Permeability. Circ. 2014, 4, 535–551, doi:10.1086/677356.
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Rev. Dis. Primers 2019, 5, 18.
- Rippe, B.; Rosengren, B.-I.; Carlsson, O.; Venturoli, D. Transendothelial Transport: The Vesicle Controversy. Vasc. Res. 2002, 39, 375–390, doi:10.1159/000064521.
- Tsushima, K.; King, L.S.; Aggarwal, N.R.; De Gorordo, A.; D’Alessio, F.R.; Kubo, K. Acute Lung Injury Review. Med. 2009, 48, 621–630, doi:10.2169/internalmedicine.48.1741.
- Liu, Y.; Mu, S.; Li, X.; Liang, Y.; Wang, L.; Ma, X. Unfractionated Heparin Alleviates Sepsis-Induced Acute Lung Injury by Protecting Tight Junctions. Surg. Res. 2019, 238, 175–185, doi:10.1016/j.jss.2019.01.020.
- Benatti, M.N.; Fabro, A.T.; Miranda, C.H. Endothelial glycocalyx shedding in the acute respiratory distress syndrome after flu syndrome. Intensive Care 2020, 8, 1–10, doi:10.1186/s40560-020-00488-7.
- Zhang, D.; Qi, B.-Y.; Zhu, W.W.; Huang, X.; Wang, X. Crocin alleviates lipopolysaccharide-induced acute respiratory distress syndrome by protecting against glycocalyx damage and suppressing inflammatory signaling pathways. Res. 2020, 69, 267–278, doi:10.1007/s00011-019-01314-z.
- Epstein, F.H.; Vane, J.R.; Änggård, E.E.; Botting, R.M. Regulatory Functions of the Vascular Endothelium. Engl. J. Med. 1990, 323, 27–36, doi:10.1056/nejm199007053230106.
- Moloney, E.D.; Evans, T.W. Pathophysiology and pharmacological treatment of pulmonary hypertension in acute respiratory distress syndrome. Respir. J. 2003, 21, 720–727, doi:10.1183/09031936.03.00120102.
- Druml, W.; Steltzer, H.; Waldhäusl, W.; Lenz, K.; Hammerle, A.; Vierhapper, H.; Gasic, S.; Wagner, O.F. Endothelin-1 in Adult Respiratory Distress Syndrome. Rev. Respir. Dis. 1993, 148, 1169–1173, doi:10.1164/ajrccm/148.5.1169.
- Sanai, L.; Haynes, W.G.; MacKenzie, A.; Grant, I.S.; Webb, D.J. Endothelin production in sepsis and the adult respiratory distress syndrome. Intensive Care Med. 1996, 22, 52–56, doi:10.1007/bf01728331.
- Langleben, D.; DeMarchie, M.; Laporta, D.; Spanier, A.H.; Schlesinger, R.D.; Stewart, D.J. Endothelin-1 in acute lung injury and the adult respiratory distress syndrome. Rev. Respir. Dis. 1993, 148 Pt 1, 1646–1650.
- Mitaka, C.; Hirata, Y.; Nagura, T.; Tsunoda, Y.; Amaha, K. Circulating Endothelin-1 Concentrations in Acute Respiratory Failure. Chest 1993, 104, 476–480, doi:10.1378/chest.104.2.476.
- Albertine, K.H.; Wang, Z.M.; Michael, J.R. Expression of endothelial nitric oxide synthase, inducible nitric oxide synthase, and endothelin-1 in lungs of subjects who died with ARDS. Chest 1999, 116 (Suppl. 1), 101S–102S.
- Nakano, Y.; Tasaka, S.; Saito, F.; Yamada, W.; Shiraishi, Y.; Ogawa, Y.; Koh, H.; Hasegawa, N.; Fujishima, S.; Hashimoto, S.; et al. Endothelin-1 level in epithelial lining fluid of patients with acute respiratory distress syndrome. Respirology 2007, 12, 740–743, doi:10.1111/j.1440-1843.2007.01115.x.
- Pritze, S.; Peskar, B.A.; Simmet, T. Release of eicosanoids and endothelin in an experimental model of adult respiratory distress syndrome. Agents Actions Suppl. 1992, 37, 41–46.
- Lai, T.-S.; Cai, S.-X.; Guo, Z.-H. Serum and lung endothelin-1 increased in a canine model of ventilator-induced lung injury. Med J. 2010, 123, 1021–1027.
- Simmet, T.; Pritze, S.; Thelen, K.I.; Peskar, B.A. Release of endothelin in the oleic acid-induced respiratory distress syndrome in rats. J. Pharmacol. 1992, 211, 319–322, doi:10.1016/0014-2999(92)90387-j.
- McCarter, S.D.; Lai, P.F.H.; Suen, R.S.; Stewart, D.J. Regulation of endothelin-1 by angiopoietin-1: Implications for inflammation. Biol. Med. 2006, 231, 985–991.
- Bhavsar, T.M.; Cerreta, J.M.; Liu, M.; Reznik, S.E.; Cantor, J.O. Phosphoramidon, an endothelin-converting enzyme inhibitor, attenuates lipopolysaccharide-induced acute lung injury. Lung Res. 2008, 34, 141–154.
- Kuklin, V.; Kirov, M.; Sovershaev, M.; Andreasen, T.; Ingebretsen, O.C.; Ytrehus, K.; Bjertnaes, L.J. Tezosentan-induced attenuation of lung injury in endotoxemic sheep is associated with reduced activation of protein kinase C. Care 2005, 9, R211–R217, doi:10.1186/cc3497.
- Atalay, F.; Yurdakan, G.; Yilmaz-Sipahi, E. Effect of the endothelin receptor antagonist tezosentan on alpha-naphthylthiourea-induced lung injury in rats. Kaohsiung J. Med. Sci. 2012, 28, 72–78, doi:10.1016/j.kjms.2011.10.019.
- Fujii, Y.; Magder, S.; Cernacek, P.; Goldberg, P.; Guo, Y.; Hussain, S.N.A. Endothelin Receptor Blockade Attenuates Lipopolysaccharide-induced Pulmonary Nitric Oxide Production. J. Respir. Crit. Care Med. 2000, 161 Pt 1, 982–989, doi:10.1164/ajrccm.161.3.9904094.
- Manitsopoulos, N.; Nikitopoulou, I.; Maniatis, N.A.; Magkou, C.; Kotanidou, A.; Orfanos, S.E. Highly Selective Endothelin-1 Receptor A Inhibition Prevents Bleomycin-Induced Pulmonary Inflammation and Fibrosis in Mice. Respiration 2018, 95, 122–136.
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Res. 2007, 100, 174–190.
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (Covid-19)? Med. 2020, 7, 335.
- Chambers, S.; Bhatia, M. ACE and ACE2 in Inflammation: A Tale of Two Enzymes. Allergy Drug Targets 2014, 13, 224–234, doi:10.2174/1871528113666140713164506.
- Casey, L.; Krieger, B.; Kohler, J.; Rice, C.; Oparil, S.; Szidon, P. Decreased serum angiotensin converting enzyme in adult respiratory distress syndrome associated with sepsis. Care Med. 1981, 9, 651–654, doi:10.1097/00003246-198109000-00008.
- Fourrier, F.; Chopi, C.; Wallaert, B.; Mazurier, C.; Mangalahoyi, J.; Durocher, A. Compared Evolution of Plasma Fibronectin and Angiotensin-converting Enzyme Levels in Septic ARDS. Chest 1985, 87, 191–195, doi:10.1378/chest.87.2.191.
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096, doi:10.1371/journal.pone.0213096.
- Idell, S.; Kueppers, F.; Lippmann, M.; Rosen, H.; Niederman, M.; Fein, A. Angiotensin Converting Enzyme in Bronchoalveolar Lavage in ARDS. Chest 1987, 91, 52–56, doi:10.1378/chest.91.1.52.
- Kaparianos, A. Local Renin-Angiotensin II Systems, Angiotensin-Converting Enzyme and its Homologue ACE2: Their Potential Role in the Pathogenesis of Chronic Obstructive Pulmonary Diseases, Pulmonary Hypertension and Acute Respiratory Distress Syndrome. Med. Chem. 2011, 18, 3506–3515, doi:10.2174/092986711796642562.
- Takei, Y.; Yamada, M.; Saito, K.; Kameyama, Y.; Sugiura, H.; Makiguchi, T.; Fujino, N.; Koarai, A.; Toyama, H.; Saito, K.; et al. Increase in circulating ACE-positive endothelial microparticles during acute lung injury. Respir. J. 2019, 54, 1801188, doi:10.1183/13993003.01188-2018.
- Yamamoto, T.; Wang, L.-M.; Shimakura, K.; Sanaka, M.; Koike, Y.; Mineshita, S. Angiotensin II-Induced Pulmonary Edema in a Rabbit Model. J. Pharmacol. 1997, 73, 33–40, doi:10.1254/jjp.73.33.
- Yilin, Z.; Yandong, N.; Faguang, J. Role of angiotensin-converting enzyme (ACE) and ACE2 in a rat model of smoke inhalation induced acute respiratory distress syndrome. Burns 2015, 41, 1468–1477, doi:10.1016/j.burns.2015.04.010.
- Hermanns, M.I.; Müller, A.M.; Tsokos, M.; Kirkpatrick, C.J. LPS-induced effects on angiotensin I-converting enzyme expression and shedding in human pulmonary microvascular endothelial cells. In Vitro Dev. Biol. Anim. 2013, 50, 287–295, doi:10.1007/s11626-013-9707-0.
- Rey-Parra, G.J.; Vadivel, A.; Coltan, L.; Hall, A.; Eaton, F.; Schuster, M.; Loibner, H.; Penninger, J.M.; Kassiri, Z.; Oudit, G.Y.; et al. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. Mol. Med. 2012, 90, 637–647, doi:10.1007/s00109-012-0859-2.
- Asperen, R.M.W.-V.; Lutter, R.; Specht, P.A.; Moll, G.N.; Van Woensel, J.B.; Van Der Loos, C.M.; Van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. Pathol. 2011, 225, 618–627, doi:10.1002/path.2987.
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Cell Biol. 2005, 436, 112–116, doi:10.1038/nature03712.
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-kappaB signaling pathways. Rep. 2016, 6, 27911.
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002, 532, 107–110, doi:10.1016/s0014-5793(02)03640-2.
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Dis. Poverty 2020, 9, 1–7, doi:10.1186/s40249-020-00662-x.
- Rigat, B.; Hubert, C.; Alhenc-Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. Clin. Investig. 1990, 86, 1343–1346, doi:10.1172/jci114844.
- Marshall, R.P.; Webb, S.; Bellingan, G.J.; Montgomery, H.E.; Chaudhari, B.; McAnulty, R.J.; Humphries, S.E.; Hill, M.R.; Laurent, G.J. Angiotensin Converting Enzyme Insertion/Deletion Polymorphism Is Associated with Susceptibility and Outcome in Acute Respiratory Distress Syndrome. J. Respir. Crit. Care Med. 2002, 166, 646–650, doi:10.1164/rccm.2108086.
- Adamzik, M.; Frey, M.U.H.; Sixt, S.; Knemeyer, L.; Beiderlinden, M.; Peters, J.; Siffert, W. ACE I/D but not AGT (-6)A/G polymorphism is a risk factor for mortality in ARDS. Respir. J. 2007, 29, 482–488, doi:10.1183/09031936.00046106.
- Matsuda, A.; Kishi, T.; Jacob, A.; Aziz, M.; Wang, P. Association between insertion/deletion polymorphism in angiotensin-converting enzyme gene and acute lung injury/acute respiratory distress syndrome: A meta-analysis. BMC Med. Genet. 2012, 13, 76, doi:10.1186/1471-2350-13-76.
- Jerng, J.-S.; Yu, C.-J.; Wang, H.-C.; Chen, K.-Y.; Cheng, S.-L.; Yang, P.-C. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Care Med. 2006, 34, 1001–1006, doi:10.1097/01.ccm.0000206107.92476.39.
- Cruces, P.; Diaz, F.; Puga, A.; Erranz, B.; Donoso, A.; Carvajal, C.; Wilhelm, J.; Repetto, G. M. Angiotensin-converting enzyme insertion/deletion polymorphism is associated with severe hypoxemia in pediatric ARDS. Intensive Care Med. 2012, 38, 113–119.
- Villar, J.; Flores, C.; Pérez-Méndez, L.; Maca-Meyer, N.; Espinosa, E.; Blanco, J.; Sangüesa, R.; Muriel, A.; Tejera, P.; Muros, M.; et al. Angiotensin-converting enzyme insertion/deletion polymorphism is not associated with susceptibility and outcome in sepsis and acute respiratory distress syndrome. Intensive Care Med. 2008, 34, 488–495, doi:10.1007/s00134-007-0937-z.
- Wang, D.; Chai, X.-Q.; Magnussen, C.G.; Zosky, G.R.; Shu, S.-H.; Wei, X.; Hu, S.-S. Renin-angiotensin-system, a potential pharmacological candidate, in acute respiratory distress syndrome during mechanical ventilation. Pharmacol. Ther. 2019, 58, 101833, doi:10.1016/j.pupt.2019.101833.
- Zhang, H.; Baker, A. Recombinant human ACE2: Acing out angiotensin II in ARDS therapy. Care 2017, 21, 1–3, doi:10.1186/s13054-017-1882-z.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Med. 2005, 11, 875–879, doi:10.1038/nm1267.
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Commun. 2014, 5, 3594, doi:10.1038/ncomms4594.

