Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Harald Wajant | -- | 2870 | 2022-11-02 08:56:38 | | | |
| 2 | Amina Yu | -2 word(s) | 2868 | 2022-11-03 02:25:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lang, I.; Zaitseva, O.; Wajant, H. CD40 as Therapeutic Target. Encyclopedia. Available online: https://encyclopedia.pub/entry/32469 (accessed on 24 July 2026).
Lang I, Zaitseva O, Wajant H. CD40 as Therapeutic Target. Encyclopedia. Available at: https://encyclopedia.pub/entry/32469. Accessed July 24, 2026.
Lang, Isabell, Olena Zaitseva, Harald Wajant. "CD40 as Therapeutic Target" Encyclopedia, https://encyclopedia.pub/entry/32469 (accessed July 24, 2026).
Lang, I., Zaitseva, O., & Wajant, H. (2022, November 02). CD40 as Therapeutic Target. In Encyclopedia. https://encyclopedia.pub/entry/32469
Lang, Isabell, et al. "CD40 as Therapeutic Target." Encyclopedia. Web. 02 November, 2022.
Copy Citation
Targeting of CD40 with the aim to stimulate or inhibit this receptor attracts considerable translational interest. Inhibitory CD40 targeting appears particularly attractive in the field of organ transplantation and in the treatment of autoimmune diseases. CD40 blockade might also elicit antitumoral activity on CD40-expressing tumors. Agonistic CD40 targeting typically aims at the exploitation of the strong immunostimulatory activities of CD40 for tumor immunotherapy and vaccination against various infectious pathogens.
antibody fusion protein
CD40
CD40L
cytokine storm
FcγR receptor
1. Inhibitory Antibody Targeting of CD40
Inhibition of CD40L-CD40 interaction can be achieved straightforwardly by conventional blocking antibodies against CD40L or CD40 (Figure 1). The important point, which has to be considered here, is to prevent binding to FcγRs and the complement activating C1q protein. The interaction with FcγRs can trigger unwanted FcγR-mediated effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC) or antibody-dependent cellular phagocytosis (ADCP) and C1q binding can elicit complement-dependent cytotoxicity (CDC) (Figure 1).
Figure 1. Isotype and the effect on CD40L-CD40 interaction determine the possible mode of actions of anti-CD40 antibodies. (A,B) Irrespective of the effect on CD40L-CD40 interaction, anti-CD40 antibodies of the appropriate isotype can stimulate inhibitory (A) or activating FcγRs (B) but also complement-mediated cell lysis via C1q binding and formation of the membrane attack complex (MAC). (B) FcγR binding by anti-CD40 antibodies further results typically in strong CD40 engagement. (C,D) Non-blocking and sensitizing anti-CD40 antibodies have no modulating effect on memCD40L-induced CD40 activation (C) while blocking antibodies completely prevent CD40 engagement by CD40L (D). (E,F) Sensitizing, non-blocking anti-CD40 antibodies enhance the activity of soluble CD40L, while non-blocking antibodies leave the weak CD40 signaling triggered by sCD40L intact. CD40 SC, CD40 signaling complex.
For example, thromboembolic complications have been reported in rhesus and cynomolgus monkeys with the mouse anti-CD40L IgG2a 5C8 and the human recombinant anti-CD40L IgG1 ABI793, and early clinical trials with the anti-CD40L antibodies Ruplizumab (BG9588, humanized 5C8) and IDEC-131 (humanized IgG1, parental antibody 24–31) were terminated due to thromboembolic events observed in a phase II study with Ruplizumab [1][2][3][4][5][6],
2. Stimulatory Antibody Targeting of CD40
Work has been ongoing for over 20 years to develop CD40 agonists with the aim to use them as adjuvants to push vaccination against various pathogens and/or to treat tumor diseases. However, these efforts have not yet resulted in approved, clinically widely applicable CD40 agonists. This failure can mainly be attributed to three circumstances/reasons: (i) the sole use of CD40 agonists as monotherapy, (ii) the dose-limiting side effects of CD40 agonists and/or FcγR- and C1q binding and (iii) the insufficient activity of the CD40 agonists used.
In preclinical animal models, the sole treatment of tumors with CD40 agonists often showed very good efficacy. However, corresponding early clinical studies with CD40 agonists were not very successful and could not prove a broad therapeutic efficacy [7]. It is now assumed that in the clinic the antitumoral efficacy of CD40 agonists is dependent on a proinflammatory microenvironment, which is typically not present in advanced tumor stages. In line with this, animal studies have shown in recent years that checkpoint inhibitors or chemotherapeutic agents that promote proinflammatory processes in the tumor microenvironment have a synergistic antitumor effect with CD40 agonists [8][9]. Therefore, in the majority of the currently ongoing clinical studies with CD40 agonists, corresponding combination therapies are being investigated [8][9].
Preclinical studies in mice have identified the cytokine release syndrome (CRS) and hepatotoxicity as two major causes of anti-CD40 antibody-induced toxicity [10][11][12]. In accordance with this, the most common side effects observed in clinical trials with CD40 agonists were symptoms of the cytokine release syndrome such as fever, nausea, muscle pain and chilling, which were transient and manageable, but also release of liver enzymes and increased lipase levels [8][9]. The various clinical studies with anti-CD40 antibodies aiming at the activation of CD40 used conventional antibodies or sometimes antibodies with preference for the binding of certain FcγR types. Importantly, clinical studies showed that anti-CD40 antibodies lacking Fc effector functions are much better tolerated than FcγR-binding competent antibody variants. For example, the anti-CD40-IgG1 antibody Lucatumumab showed in clinical trials a maximum tolerated dose (MTD) between 3 and 4.5 mg/kg with grade 3/4 adverse effects in 32–62% of the treated patients, while its Fc-silent anti-CD40-IgG1(N297A) variant Iscalimab was well tolerated up to doses of 30 mg/kg [13][14][15][16]. The higher toxicity of FcγR-binding competent anti-CD40 antibodies clearly indicates that the FcγR-bound anti-CD40 antibodies rather than the free anti-CD40 antibody molecules are the origin of the dose-limiting activities observed in clinical trials with anti-CD40 antibodies. As discussed below in detail (see Section 2.2.1), CD40 is typically much stronger activated by FcγR-interacting anti-CD40 antibodies than by antibody molecules without FcγR binding competence. Therefore, it is difficult to attribute the dose-limiting toxicity of FcγR-/C1q-binding competent anti-CD40 antibodies to the engagement of CD40 and/or FcγR signaling and complement activation. It is thus currently unclear to which extent highly potent effector function-dead agonistic CD40 antibodies trigger the aforementioned dose-limiting effects, too. However, the preclinical studies mentioned above identified typical CD40-induced effector molecules, such as IL-12p40, TNF and IFNγ, as mediators of the toxic effects of FcγR-interacting anti-CD40 antibodies [10][12]. This suggests that CD40 signaling indeed substantially contributes to the dose-limiting toxicity of FcγR-/C1q-binding competent anti-CD40 antibodies. Therefore, it appears not unlikely that FcγR-independent CD40 agonists will also elicit dose-limiting toxicity upon systemic application thereby preventing the maximal possible CD40 activation in the tumor microenvironment. In this respect, it is worth mentioning that preclinical studies have shown that the intratumoral and systemic application of anti-CD40 antibodies is therapeutically equally effective, but that local application in the tumor is associated with fewer side effects [17][18][19][20][21]. Moreover, a recent study showed that TNF inhibition prevented the hepatotoxicity triggered by combined treatment with an anti-CD40 antibody and gemcitabine without affecting antitumor activity [22]. Thus, the maximal exploitation of potent autonomous CD40 agonists for tumor therapy may require defined treatment regimens that restrict the antibody activity to the tumor area and/or systemically inhibit CD40 effector molecules.
CD40 is strongly expressed on the surface of many hematological malignancies and CD40 expression can also be quite high on solid tumors. In line with this, early on there were also tumor therapy concepts with anti-CD40 antibodies aimed at the exploitation of Fc domain-mediated immune effector mechanisms, such as ADCC (antibody-dependent cellular cytotoxicity), CDC (complement dependent cytotoxicity) and ADCP (antibody-dependent cellular phagocytosis) to destruct CD40-expressing tumor cells (Figure 3B). In accordance with the fact, discussed below in detail, that FcγR-interacting anti-CD40 antibodies regularly acquire potent CD40-stimulatory activity (Figure 3A,B), there is furthermore evidence that such immune effector function-stimulating antibodies also trigger cell death and growth arrest by CD40 engagement. Indeed, proapoptotic CD40 effects have been described in various tumor entities [23] but antiapoptotic CD40 activities have been reported as well (e.g., [24][25]). In view of the fact that CD40 signals proliferation of non-transformed B-cells [26] and protects B-cells from cell surface immunoglobulin- and CD95-induced cell death [27][28][29], the cytotoxic activity of anti-CD40 antibodies on B-cell lymphomas is at first glance counterintuitive but could reflect that the cellular vulnerability to CD40 depends from signal strength, context and differentiation status of the cell. Indeed, CD40-induced upregulation of death ligands and the death receptor CD95 along with apoptosis induction in the absence of B-cell receptor (BCR) signaling have also been reported for non-transformed B-cells [30][31][32][33][34].
The balance between the triggering of immune effector functions (ADCC, ADCP) and CD40 signaling induced by FcγR-interacting anti-CD40 antibodies in vivo is obviously not only dependent on the availability of FcγR-expressing immune cells in the neighborhood of CD40-expressing cells but also on the FcγR type expressed by these immune cells. Dominant expression of inhibitory FcγRs could favor triggering of CD40 signaling while preferential expression of activating FcγRs could tip the balance towards cell destructive immune effector mechanisms (Figure 1A,B). Accordingly, anti-CD40 antibodies with mutations conferring preference for binding of a certain FcγR type have the potential to shape the in vivo activity of anti-CD40 antibodies towards a certain direction. For example, Horton et al. [35] introduced in a humanized IgG1 variant of the anti-CD40 antibody S2C6 two point mutations (S239D/I332E) conferring, in comparison to the non-mutated IgG1 molecule, strongly enhanced binding to all human and murine FcγRs, in particular to human FcγRIIIa and murine FcγR1 and FcγRIV. The resulting antibody XmAbCD40 and its parental IgG1 variant induced with a similar dose-response antiproliferative effects in Raji and Ramos cells but XmAbCD40 showed a significantly enhanced ability to trigger ADCC and ADCP [35]. This suggests that the particular strong increase in affinity for FcγRIIIa in XmAbCD40 has preferentially affected the ability of the antibody to trigger cell destructive immune effector functions. Vice versa, anti-CD40 antibody variants specific for murine or human CD40 harboring mutations selectively enhancing the affinity for the human inhibitory antibody FcγR2B showed strongly enhanced CD40 signaling in vitro and in FcγR2B and CD40/FcγR2B humanized mice [36][37]. Remarkably, at higher doses the FcγR2B-selective human CD40 antibody showed significant hepatotoxicity hindering tumor therapy by systemic application, but the latter could be overcome by intratumoral injection [19].
The in vivo effects of some anti-CD40 antibodies stimulating immune effector mechanisms could be further complicated by the fact that these antibodies also interfere with CD40L-CD40 interaction or modulate the activity of soluble CD40L. Therefore, at localizations where neither stimulatory nor inhibitory FcγRs are present/available they might neither destruct CD40-expressing cells nor stimulate CD40 signaling but instead block CD40 engagement by endogenous CD40L or enhance the activity of soluble CD40L (Figure 1D,E and Figure 2A). For example, the anti-CD40-IgG1 Lucatumumab (HCD122, CHIR-12.12) triggers antibody-dependent cell-mediated cytotoxicity (ADCC) but also efficiently inhibits CD40L-CD40 interaction [38][39]. While attempts to target lymphoma with this antibody aimed on the exploitation of both of these functions, a Fc-silenced form of this antibody (CFZ533) was generated to avoid ADCC and to solely block CD40-CD40L interaction for immunosuppressive treatments [40]. Indeed, as already discussed., CFZ533 has been successfully used in nonhuman primates to prolong renal allograft survival without inducing B-cell depletion and Iscalimab, a fully humanized version of CFZ533, showed clinical activity in patients suffering on Graves Disease in a proof-of-concept trial and was well tolerated in a phase I study (NCT02089087) with healthy subjects and rheumatoid arthritis patients [13][41][42]. Furthermore, certain non-blocking anti-CD40 antibodies might be able to induce clustering of poorly active sCD40L-induced CD40 complexes resulting in enhanced CD40 signaling [43][44][45][46]; (Figure 1E and Figure 2A).
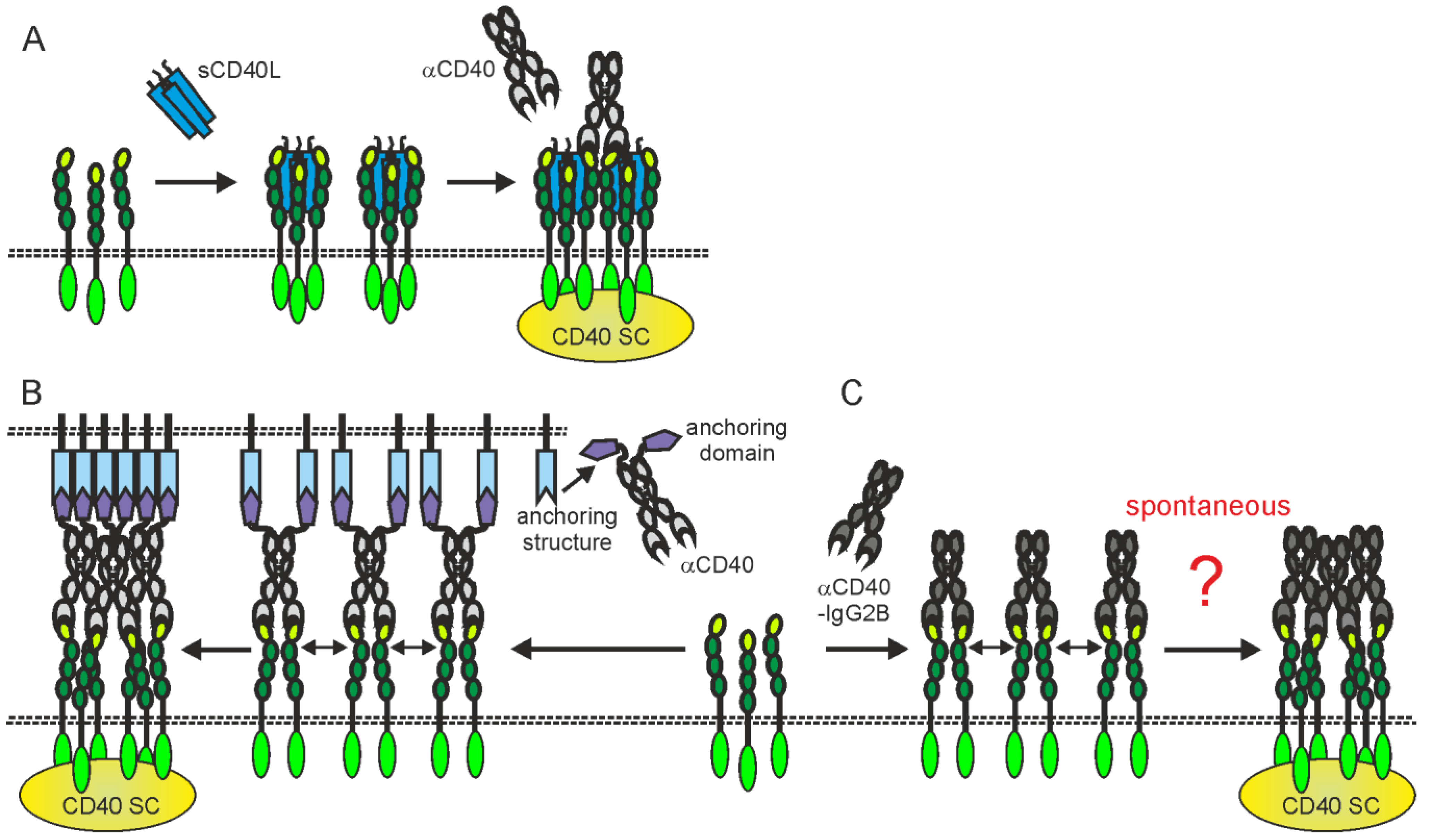
Figure 2. Activating CD40 clustering by (A) mixtures of sCD40L and non-blocking anti-CD40 antibodies, (B) anti-CD40 antibody fusion proteins with an anchoring domain enabling binding to a plasma membrane-localized anchoring structure or (C) anti-CD40-hIgG2 antibodies. Dotted lines indicate plasma membranes. For details, please see text.
2.1. Agonism of Complexes of Anti-CD40 Antibodies and FcγRs
With respect to anti-CD40 antibodies, it is extremely important to distinguish between FcγR-independent and FcγR-dependent agonistic activity, thus between the intrinsic ability of an antibody alone to trigger CD40 signaling and the ability of complexes of an antibody with FcγRs to do so. In the past 10 years, extensive in vitro and in vivo studies have given comprehensive evidence that virtually every CD40-specific antibody elicits agonistic activity when bound to Fcγ receptors [36][37][47][48]. It is worth mentioning that the agonism of FcγR-interacting anti-CD40 antibodies is independent of FcγR downstream signaling [37] and can also be realized with FcγR-transfected non-immune cells e.g., [48][49][50]. Therefore, the sheer plasma membrane-associated mode of presentation of anti-CD40 antibody molecules appears to be sufficient to constitute the agonism of anti-CD40 antibody-FcγR complexes. The nature of the FcγR type appears only to be in so far of relevance for the agonism of anti-CD40 antibodies that FcγRs differ in their affinity for the various IgG isotypes and that therefore certain anti-CD40-IgG-FcγR complexes form more efficiently than others. This issue, however, can gain overwhelming importance in vivo since the type of immune cell present in a certain tumor entity as well as the FcγR expression pattern of the various immune cell types varies considerably and the different antibody isotypes have quite different preferences for FcγRs [51][52]. Therefore, the combination of the availability of the “right” immune cell type together with the FcγR specificity of a certain anti-CD40 antibody isotype has obviously a significant impact on the achievable agonism in vivo and can explain why anti-CD40 antibodies show quite different in vivo performance ranging from antagonism over model-dependent quantitatively widely differing agonism despite having a comparable FcγR-dependent agonistic activity in vitro.
Several groups have shown that bispecific anti-CD40 antibody variants that recognize plasma membrane-associated targets distinct from CD40 elicit up to a 1000-fold increased CD40-stimulating activity after binding to this second antigen [50][53][54][55][56]. These studies not only demonstrate that the agonism manifesting anti-CD40 antibody-FcγR interaction can be replaced by molecularly different interactions emphasizing the relevance of the plasma membrane-associated presentation mode for agonism, but also offers the opportunity to prevent systemic CD40 activation by addressing a selectively expressed target, e.g., a tumor antigen.
It appears quite plausible that the agonism of FcγR binding-competent anti-CD40 antibodies is due to the same molecular mode of action that also applies to the much stronger CD40-stimulating activity of membrane CD40L compared to soluble CD40L trimers. Namely, the presence of high local concentrations of plasma membrane agonist-bound CD40 molecules (FcγR-anti-CD40-antibody-CD40 dimers) in the cell–cell contact zone between CD40+ cells and FcγR+ cells favoring secondary clustering to fully active oligomeric agonist-CD40 complexes.
In view of the fact that anti-CD40 antibodies bound to FcγRs regularly display strong agonism, it is evident that the clinical development of in vivo antagonistically acting anti-CD40 antibodies is de facto only possible with antibody isotypes that do not or only very slightly interact with FcγRs, or with immunoglobulin mutants with defective FcγR binding (e.g., IgG1-N297A or IgG1-LALA).
2.2. Problems and Limitations of CD40 Engagement by FcγR-Interacting Anti-CD40 Antibodies
In general, it must be considered that it is typically not possible to achieve activation of all CD40 molecules with anti-CD40 antibodies in vivo, due to the limited availability of FcγR molecules. For example, anti-CD40-mIgG1 antibodies stimulate significant proliferation of B-cells from wild-type mice but not of B-cells from FcγRIIB-deficient mice [48]. However, this FcγRIIB-dependent CD40 agonism can be further increased by one to two orders of magnitude in the wild-type and FcγRIIB-deficient B-cells if FcγR-expressing transfectants are added [48]. Apparently, the physiological FcγR expression levels of B-cells are not sufficient to allow occupancy of all CD40 molecules of the B-cells with FcγR binding-competent anti-CD40 antibody molecules. Furthermore, the binding of anti-CD40 antibodies to activating FcγRs not only empower these antibodies to efficiently activate CD40 but, as discussed already before, can also result in the destruction of the CD40-expressing target cells by effector functions of the FcγR-expressing cells. Finally, conventional anti-CD40 antibodies have to compete with endogenous IgG molecules for FcγR binding, resulting in the need to apply high anti-CD40 antibody doses to reach relevant FcγR occupation.
2.3. Anti-CD40 Antibodies with Intrinsic Thus FcγR-Independent Agonism
The majority of reports on agonistic anti-CD40 antibodies investigated CD40 agonism in FcγR-expressing cell types (DCs, B-cells) or observed enhanced anti-CD40 agonism upon crosslinking with secondary antibodies but nevertheless imprecisely attributed the agonism solely to the anti-CD40 antibody and not to the FcγR-bound antibodies or the anti-CD40-anti-IgG complexes. Thus, many published “agonistic” anti-CD40 antibodies have no or only extremely moderate intrinsic agonistic activity.
However, some studies have explicitly demonstrated robust intrinsic autonomous agonism of anti-CD40 antibodies, especially for anti-CD40 antibodies of the human IgG2 isotype (hIgG2) [57]. Interestingly, the agonistic activity of anti-CD40-hIgG2 antibodies has been assigned to isoform B of the hIgG2 isotype, which differs from the A isoform of the hIgG2 molecule in the formation of disulfide bridges between the CH1 and CL domains, and has a less flexible arrangement of the two Fab domains of the antibody [58][59][60][61]. Anti-CD40-hIgG2 antibodies that have mutations that produce either only isoform A (e.g., HC-C233S) or only isoform B (e.g., HC-C127S or LC-C214S/HC-C233S) therefore elicit no agonistic activity or even show an increased FcγR-independent agonism compared to the parental hIgG2 molecule [57]. In line with the two-step model of CD40 activation, it has been found that most anti-CD40-IgG2B antibodies, in contrast to their IgG1 counterparts, indeed autonomously trigger strong CD40 clustering [62]. However, it is unclear, whether the secondary clustering of initially formed CD40-IgG2B complexes is powered by CD40-CD40 or IgG2B-IgG2B interactions (Figure 2C). It is also worth mentioning that the FcγR-independent agonism of anti-CD40-hIgG2 or anti-CD40-hIgG2B antibodies seems to still be significantly lower than that of FcγR binding-competent anti-CD40 antibodies in the presence of FcγRs or have been challenged for its relevance in the human system [36][48].
References
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727.
- Brams, P.; Black, A.; Padlan, E.A.; Hariharan, K.; Leonard, J.; Chambers-Slater, K.; Noelle, R.J.; Newman, R. A humanized anti-human CD154 monoclonal antibody blocks CD154-CD40 mediated human B cell activation. Int. Immunopharmacol. 2001, 1, 277–294.
- Kalunian, K.C.; Davis, J.C., Jr.; Merrill, J.T.; Totoritis, M.C.; Wofsy, D. Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 3251–3258.
- Kawai, T.; Andrews, D.; Colvin, R.B.; Sachs, D.H.; Cosimi, A.B. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat. Med. 2000, 6, 114.
- Koyama, I.; Kawai, T.; Andrews, D.; Boskovic, S.; Nadazdin, O.; Wee, S.L.; Sogawa, H.; Wu, D.L.; Smith, R.N.; Colvin, R.B.; et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation 2004, 77, 460–462.
- Schuler, W.; Bigaud, M.; Brinkmann, V.; Di Padova, F.; Geisse, S.; Gram, H.; Hungerford, V.; Kleuser, B.; Kristofic, C.; Menninger, K.; et al. Efficacy and safety of ABI793, a novel human anti-human CD154 monoclonal antibody, in cynomolgus monkey renal allotransplantation. Transplantation 2004, 77, 717–726.
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013, 19, 1035–1043.
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58.
- Li, D.K.; Wang, W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol. Lett. 2020, 20, 176.
- Bonnans, C.; Thomas, G.; He, W.; Jung, B.; Chen, W.; Liao, M.; Heyen, J.; Buetow, B.; Pillai, S.; Matsumoto, D.; et al. CD40 agonist-induced IL-12p40 potentiates hepatotoxicity. J. Immunother. Cancer 2020, 8, e000624.
- Medina-Echeverz, J.; Ma, C.; Duffy, A.G.; Eggert, T.; Hawk, N.; Kleiner, D.E.; Korangy, F.; Greten, T.F. Systemic Agonistic Anti-CD40 Treatment of Tumor-Bearing Mice Modulates Hepatic Myeloid-Suppressive Cells and Causes Immune-Mediated Liver Damage. Cancer Immunol. Res. 2015, 3, 557–566.
- Siwicki, M.; Gort-Freitas, N.A.; Messemaker, M.; Bill, R.; Gungabeesoon, J.; Engblom, C.; Zilionis, R.; Garris, C.; Gerhard, G.M.; Kohl, A.; et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci. Immunol. 2021, 6, eabi7083.
- Espié, P.; He, Y.; Koo, P.; Sickert, D.; Dupuy, C.; Chokoté, E.; Schuler, R.; Mergentaler, H.; Ristov, J.; Milojevic, J.; et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am. J. Transplant. 2020, 20, 463–473.
- Bensinger, W.; Maziarz, R.T.; Jagannath, S.; Spencer, A.; Durrant, S.; Becker, P.S.; Ewald, B.; Bilic, S.; Rediske, J.; Baeck, J.; et al. A phase 1 study of lucatumumab, a fully human anti-CD40 antagonist monoclonal antibody administered intravenously to patients with relapsed or refractory multiple myeloma. Br. J. Haematol. 2012, 159, 58–66.
- Byrd, J.C.; Kipps, T.J.; Flinn, I.W.; Cooper, M.; Odenike, O.; Bendiske, J.; Rediske, J.; Bilic, S.; Dey, J.; Baeck, J.; et al. Phase I study of the anti-CD40 humanized monoclonal antibody lucatumumab (HCD122) in relapsed chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 2136–2142.
- Fanale, M.; Assouline, S.; Kuruvilla, J.; Solal-Céligny, P.; Heo, D.S.; Verhoef, G.; Corradini, P.; Abramson, J.S.; Offner, F.; Engert, A.; et al. Phase IA/II, multicentre, open-label study of the CD40 antagonistic monoclonal antibody lucatumumab in adult patients with advanced non-Hodgkin or Hodgkin lymphoma. Br. J. Haematol. 2014, 164, 258–265.
- Fransen, M.F.; Sluijter, M.; Morreau, H.; Arens, R.; Melief, C.J. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin. Cancer Res. 2011, 17, 2270–2280.
- Jackaman, C.; Lew, A.M.; Zhan, Y.; Allan, J.E.; Koloska, B.; Graham, P.T.; Robinson, B.W.; Nelson, D.J. Deliberately provoking local inflammation drives tumors to become their own protective vaccine site. Int. Immunol. 2008, 20, 1467–1479.
- Knorr, D.A.; Dahan, R.; Ravetch, J.V. Toxicity of an Fc-engineered anti-CD40 antibody is abrogated by intratumoral injection and results in durable antitumor immunity. Proc. Natl. Acad. Sci. USA 2018, 115, 11048–11053.
- Sandin, L.C.; Orlova, A.; Gustafsson, E.; Ellmark, P.; Tolmachev, V.; Tötterman, T.H.; Mangsbo, S.M. Locally delivered CD40 agonist antibody accumulates in secondary lymphoid organs and eradicates experimental disseminated bladder cancer. Cancer Immunol. Res. 2014, 2, 80–90.
- van Mierlo, G.J.; den Boer, A.T.; Medema, J.P.; van der Voort, E.I.; Fransen, M.F.; Offringa, R.; Melief, C.J.; Toes, R.E. CD40 stimulation leads to effective therapy of CD40(−) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc. Natl. Acad. Sci. USA 2002, 99, 5561–5566.
- Stone, M.L.; Lee, J.; Herrera, V.M.; Graham, K.; Lee, J.W.; Huffman, A.; Coho, H.; Tooker, E.; Myers, M.I.; Giannone, M.; et al. TNF blockade uncouples toxicity from antitumor efficacy induced with CD40 chemoimmunotherapy. JCI Insight 2021, 6, e146314.
- Geldart, T.; Illidge, T. Anti-CD 40 monoclonal antibody. Leuk. Lymphoma 2005, 46, 1105–1113.
- Andersen, N.S.; Larsen, J.K.; Christiansen, J.; Pedersen, L.B.; Christophersen, N.S.; Geisler, C.H.; Jurlander, J. Soluble CD40 ligand induces selective proliferation of lymphoma cells in primary mantle cell lymphoma cell cultures. Blood 2000, 96, 2219–2225.
- Ghia, P.; Boussiotis, V.A.; Schultze, J.L.; Cardoso, A.A.; Dorfman, D.M.; Gribben, J.G.; Freedman, A.S.; Nadler, L.M. Unbalanced expression of bcl-2 family proteins in follicular lymphoma: Contribution of CD40 signaling in promoting survival. Blood 1998, 91, 244–251.
- Bishop, G.A.; Hostager, B.S. Signaling by CD40 and its mimics in B cell activation. Immunol. Res. 2001, 24, 97–109.
- Cleary, A.M.; Fortune, S.M.; Yellin, M.J.; Chess, L.; Lederman, S. Opposing roles of CD95 (Fas/APO-1) and CD40 in the death and rescue of human low density tonsillar B cells. J. Immunol. 1995, 155, 3329–3337.
- Lagresle, C.; Mondière, P.; Bella, C.; Krammer, P.H.; Defrance, T. Concurrent engagement of CD40 and the antigen receptor protects naive and memory human B cells from APO-1/Fas-mediated apoptosis. J. Exp. Med. 1996, 183, 1377–1388.
- Tsubata, T.; Wu, J.; Honjo, T. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature 1993, 364, 645–648.
- Eliopoulos, A.G.; Davies, C.; Knox, P.G.; Gallagher, N.J.; Afford, S.C.; Adams, D.H.; Young, L.S. CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol. Cell Biol. 2000, 20, 5503–5515.
- Garrone, P.; Neidhardt, E.M.; Garcia, E.; Galibert, L.; van Kooten, C.; Banchereau, J. Fas ligation induces apoptosis of CD40-activated human B lymphocytes. J. Exp. Med. 1995, 182, 1265–1273.
- Lagresle, C.; Bella, C.; Daniel, P.T.; Krammer, P.H.; Defrance, T. Regulation of germinal center B cell differentiation. Role of the human APO-1/Fas (CD95) molecule. J. Immunol. 1995, 154, 5746–5756.
- Ribeiro, P.; Renard, N.; Warzocha, K.; Charlot, C.; Jeandenant, L.; Callet-Bauchu, E.; Coiffier, B.; Salles, G. CD40 regulation of death domains containing receptors and their ligands on lymphoma B cells. Br. J. Haematol. 1998, 103, 684–689.
- Schattner, E.J.; Elkon, K.B.; Yoo, D.H.; Tumang, J.; Krammer, P.H.; Crow, M.K.; Friedman, S.M. CD40 ligation induces Apo-1/Fas expression on human B lymphocytes and facilitates apoptosis through the Apo-1/Fas pathway. J. Exp. Med. 1995, 182, 1557–1565.
- Horton, H.M.; Bernett, M.J.; Peipp, M.; Pong, E.; Karki, S.; Chu, S.Y.; Richards, J.O.; Chen, H.; Repp, R.; Desjarlais, J.R.; et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood 2010, 116, 3004–3012.
- Dahan, R.; Barnhart, B.C.; Li, F.; Yamniuk, A.P.; Korman, A.J.; Ravetch, J.V. Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcγR Engagement. Cancer Cell 2016, 29, 820–831.
- Li, F.; Ravetch, J.V. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 2011, 333, 1030–1034.
- Tai, Y.T.; Li, X.; Tong, X.; Santos, D.; Otsuki, T.; Catley, L.; Tournilhac, O.; Podar, K.; Hideshima, T.; Schlossman, R.; et al. Human anti-CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res. 2005, 65, 5898–5906.
- Luqman, M.; Klabunde, S.; Lin, K.; Georgakis, G.V.; Cherukuri, A.; Holash, J.; Goldbeck, C.; Xu, X.; Kadel, E.E., 3rd; Lee, S.H.; et al. The antileukemia activity of a human anti-CD40 antagonist antibody, HCD122, on human chronic lymphocytic leukemia cells. Blood 2008, 112, 711–720.
- Ristov, J.; Espie, P.; Ulrich, P.; Sickert, D.; Flandre, T.; Dimitrova, M.; Müller-Ristig, D.; Weider, D.; Robert, G.; Schmutz, P.; et al. Characterization of the in vitro and in vivo properties of CFZ533, a blocking and non-depleting anti-CD40 monoclonal antibody. Am. J. Transplant. 2018, 18, 2895–2904.
- Cordoba, F.; Wieczorek, G.; Audet, M.; Roth, L.; Schneider, M.A.; Kunkler, A.; Stuber, N.; Erard, M.; Ceci, M.; Baumgartner, R.; et al. A novel, blocking, Fc-silent anti-CD40 monoclonal antibody prolongs nonhuman primate renal allograft survival in the absence of B cell depletion. Am. J. Transplant. 2015, 15, 2825–2836.
- Kahaly, G.J.; Stan, M.N.; Frommer, L.; Gergely, P.; Colin, L.; Amer, A.; Schuhmann, I.; Espie, P.; Rush, J.S.; Basson, C.; et al. A Novel Anti-CD40 Monoclonal Antibody, Iscalimab, for Control of Graves Hyperthyroidism-A Proof-of-Concept Trial. J. Clin. Endocrinol. Metab. 2020, 105, 696–704.
- Yu, X.; Chan, H.T.C.; Fisher, H.; Penfold, C.A.; Kim, J.; Inzhelevskaya, T.; Mockridge, C.I.; French, R.R.; Duriez, P.J.; Douglas, L.R.; et al. Isotype Switching Converts Anti-CD40 Antagonism to Agonism to Elicit Potent Antitumor Activity. Cancer Cell 2020, 37, 850–866.e7.
- Wu, H.H.; Ralph, K.L.; Sepuldeva, E.; Hansen, G.; Li, H.; Huang, Z.F.; Liu, D.; Dziegelewski, M.; Ahlberg, J.; Frego, L.; et al. An optimally designed anti-human CD40 antibody with potent B cell suppression for the treatment of autoimmune diseases. Int. J. Pharm. 2021, 609, 121162.
- Ceglia, V.; Zurawski, S.; Montes, M.; Kroll, M.; Bouteau, A.; Wang, Z.; Ellis, J.; Igyártó, B.Z.; Lévy, Y.; Zurawski, G. Anti-CD40 Antibody Fused to CD40 Ligand Is a Superagonist Platform for Adjuvant Intrinsic DC-Targeting Vaccines. Front. Immunol. 2021, 12, 786144.
- Schwabe, R.F.; Hess, S.; Johnson, J.P.; Engelmann, H. Modulation of soluble CD40 ligand bioactivity with anti-CD40 antibodies. Hybridoma 1997, 16, 217–226.
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011, 19, 101–113.
- Yu, X.; Chan, H.T.C.; Orr, C.M.; Dadas, O.; Booth, S.G.; Dahal, L.N.; Penfold, C.A.; O’Brien, L.; Mockridge, C.I.; French, R.R.; et al. Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell 2018, 33, 664–675.e4.
- Filbert, E.L.; Björck, P.K.; Srivastava, M.K.; Bahjat, F.R.; Yang, X. APX005M, a CD40 agonist antibody with unique epitope specificity and Fc receptor binding profile for optimal therapeutic application. Cancer Immunol. Immunother. 2021, 70, 1853–1865.
- Nelke, J.; Medler, J.; Weisenberger, D.; Beilhack, A.; Wajant, H. CD40- and CD95-specific antibody single chain-Baff fusion proteins display BaffR-, TACI- and BCMA-restricted agonism. MAbs 2020, 12, 1807721.
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643.
- Nimmerjahn, F.; Gordan, S.; Lux, A. FcγR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol. 2015, 36, 325–336.
- Medler, J.; Kucka, K.; Melo, V.; Zhang, T.; von Rotenhan, S.; Ulrich, J.; Bremer, E.; Hudecek, M.; Beilhack, A.; Wajant, H. CD40- and 41BB-specific antibody fusion proteins with PDL1 blockade-restricted agonism. Theranostics 2022, 12, 1486–1499.
- Medler, J.; Nelke, J.; Weisenberger, D.; Steinfatt, T.; Rothaug, M.; Berr, S.; Hünig, T.; Beilhack, A.; Wajant, H. TNFRSF receptor-specific antibody fusion proteins with targeting controlled FcγR-independent agonistic activity. Cell Death Dis. 2019, 10, 224.
- Sum, E.; Rapp, M.; Fröbel, P.; Le Clech, M.; Dürr, H.; Giusti, A.M.; Perro, M.; Speziale, D.; Kunz, L.; Menietti, E.; et al. Fibroblast Activation Protein α-Targeted CD40 Agonism Abrogates Systemic Toxicity and Enables Administration of High Doses to Induce Effective Antitumor Immunity. Clin. Cancer Res. 2021, 27, 4036–4053.
- Ye, S.; Cohen, D.; Belmar, N.A.; Choi, D.; Tan, S.S.; Sho, M.; Akamatsu, Y.; Kim, H.; Iyer, R.; Cabel, J.; et al. A Bispecific Molecule Targeting CD40 and Tumor Antigen Mesothelin Enhances Tumor-Specific Immunity. Cancer Immunol. Res. 2019, 7, 1864–1875.
- White, A.L.; Chan, H.T.; French, R.R.; Willoughby, J.; Mockridge, C.I.; Roghanian, A.; Penfold, C.A.; Booth, S.G.; Dodhy, A.; Polak, M.E.; et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 2015, 27, 138–148.
- Dillon, T.M.; Ricci, M.S.; Vezina, C.; Flynn, G.C.; Liu, Y.D.; Rehder, D.S.; Plant, M.; Henkle, B.; Li, Y.; Deechongkit, S.; et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J. Biol. Chem. 2008, 283, 16206–16215.
- Martinez, T.; Guo, A.; Allen, M.J.; Han, M.; Pace, D.; Jones, J.; Gillespie, R.; Ketchem, R.R.; Zhang, Y.; Balland, A. Disulfide connectivity of human immunoglobulin G2 structural isoforms. Biochemistry 2008, 47, 7496–7508.
- Ryazantsev, S.; Tischenko, V.; Nguyen, C.; Abramov, V.; Zav’yalov, V. Three-dimensional structure of the human myeloma IgG2. PLoS ONE 2013, 8, e64076.
- Wypych, J.; Li, M.; Guo, A.; Zhang, Z.; Martinez, T.; Allen, M.J.; Fodor, S.; Kelner, D.N.; Flynn, G.C.; Liu, Y.D.; et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J. Biol. Chem. 2008, 283, 16194–16205.
- Yu, X.; James, S.; Felce, J.H.; Kellermayer, B.; Johnston, D.A.; Chan, H.T.C.; Penfold, C.A.; Kim, J.; Inzhelevskaya, T.; Mockridge, C.I.; et al. TNF receptor agonists induce distinct receptor clusters to mediate differential agonistic activity. Commun. Biol. 2021, 4, 772.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
03 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No