 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhaohui Feng | + 4136 word(s) | 4136 | 2020-11-18 07:09:37 | | | |
| 2 | Bruce Ren | Meta information modification | 4136 | 2020-11-27 02:55:28 | | |
Video Upload Options
Tumor suppressor p53 plays a key role in tumor suppression. In addition to tumor suppression, p53 is also involved in many other biological and pathological processes, such as immune response, maternal reproduction, tissue ischemia/reperfusion injuries and neurodegenerative diseases. While it has been widely accepted that the role of p53 in regulation of cell cycle arrest, senescence and apoptosis contributes greatly to the function of p53 in tumor suppression, emerging evidence has implicated that p53 also exerts its tumor suppressive function through regulation of many other cellular processes, such as metabolism, anti-oxidant defense and ferroptosis. Ferroptosis is a unique iron-dependent form of programmed cell death driven by lipid peroxidation in cells. Ferroptosis has been reported to be involved in cancer, tissue ischemia/reperfusion injuries and neurodegenerative diseases. Recent studies have shown that ferroptosis can be regulated by p53 and its signaling pathway as well as tumor-associated mutant p53. Interestingly, the regulation of ferroptosis by p53 appears to be highly context-dependent.
1. Introduction
Tumor suppressor p53 plays a key role in tumor suppression [1][2][3][4]. p53 function is disrupted in many human cancers through mutations of the p53 gene and other mechanisms, including amplification and/or overexpression of p53 negative regulators (e.g., MDM2 and MDM4), which is a prerequisite for the initiation and/or progression of many human cancers [1][2][3][4][5]. The p53 gene is mutated in over half of all human cancers and almost in every type of cancers [6][7][8][9]. As a transcription factor, p53 mainly exerts its tumor suppressive function through selective transcriptional regulation of many target genes to regulate various fundamental cellular responses, including apoptosis, cell cycle arrest, senescence, DNA repair and metabolism [1][2][3][4]. In addition to transcription regulation, p53 also directly interacts with other proteins to regulate different cellular responses, such as apoptosis and DNA repair [10][11]. In human cancers, majority of p53 mutations are missense mutations, which leads to the production of full-length mutant p53 (mutp53) proteins in cancer cells [5][6][7][8]. Different from wild-type p53 proteins in normal cells, mutp53 proteins frequently accumulate to very high levels in cancer cells. Notably, many missense mutp53 proteins have been demonstrated to not only lose the tumor suppressive function of wild-type p53 but also exert gain-of-function (GOF) activities to promote cancer progression independently of wild-type p53 [12][13].
Recent studies have revealed a novel function of p53 in regulation of ferroptosis, a unique iron-dependent form of cell death driven by the accumulation of lipid-based reactive oxygen species (ROS) in cells [14][15][16][17][18]. Ferroptosis is a specific form of cell death that was originally found to be induced by small molecules erastin and RSL3 (RAS-selective lethal 3), which were identified in the synthetic lethal screening for small molecules targeting cancer cells with overexpression of oncogenic RAS [19][20]. Ferroptosis has been reported to be involved in different physiological and pathological processes, including cancer, neurodegenerative diseases, tissue ischemia/reperfusion injuries and immune response [21]. Recent studies reported that the regulation of ferroptosis by p53 contributes to the tumor suppressive function of p53 and furthermore, mutp53 protein accumulation in cancer cells sensitizes cancer cells to ferroptosis [22][23][24].
2. The Regulation of Ferroptosis by p53 and its Signaling Pathway
2.1. Wild-type p53 Induces Ferroptosis
p53 was first reported to sensitize cells to ferroptosis through transcriptionally repressing expression of SLC7A11 by Wei Gu’s group [25]. SLC7A11 is a direct p53 target; p53 binds to the p53-responsive element in the promoter region of SLC7A11 to repress its expression, leading to enhanced sensitivity of cancer cells to ferroptosis inducers, such as erastin [25]. Interestingly, p533KR, an acetylation-defective p53 mutant containing mutations of three lysine residues (K117R, K161R and K162R) in the DNA-binding domain of p53, fails to induce cell cycle arrest, senescence and apoptosis but can effectively repress SLC7A11 expression and induce ferroptosis in response to ROS-induced stress [25]. Furthermore, p53-mediated ferroptosis contributes to embryonic development and the lethality induced by MDM2 knockout in mice [25]. SLC7A11 is frequently overexpressed in different types of human cancers, including colorectal, liver and kidney cancers [25][26][27]. Notably, ectopic expression of SLC7A11 inhibits ferroptosis and abolishes the tumor suppressive function of p533KR in xenograft tumor models [25]. The same group further showed that p534KR98, an acetylation-defective p53 mutant with an additional lysine 98 (K98) mutation in p533KR, fails to repress SLC711A expression or induce ferroptosis. Notably, p534KR98 results in loss of tumor suppressive function of p53; compared with p533KR mice, the p534KR98 mice are prone to tumor development [28]. These results provided one of the first lines of evidence that ferroptosis has tumor suppressive function and furthermore, the induction of ferroptosis by p53 contributes to the function of p53 in tumor suppression (Figure 1). Recently, monoubiquitination of histone H2B on lysine 120 (H2Bub1), an epigenetic mark for transcriptional activation, was reported to be involved in the regulation of SLC7A11 and ferroptosis [29]. H2Bub1 promotes SLC7A11 expression, whereas loss of H2Bub1 reduces SLC7A11 expression and leads to ferroptosis. Interestingly, p53 acts as a negative regulator of H2Bub1 by interacting with and promoting the nuclear translocation of USP7, a deubiquitinase that removes H2Bub1 from the regulatory region of the SLC7A11 gene, leading to decreased SLC7A11 expression. This p53-USP7-H2Bub1-SLC7A11 axis sensitizes cells to erastin-induced ferroptosis [29].
The low-molecular-weight polyamines, including putrescine, spermidine and spermine, are amino acid-derived polycationic alkylamines essential for cellular growth, proliferation and survival [30]. Their levels are tightly regulated by enzymes involved in polyamine metabolism, which is frequently dysregulated in different types of cancers, such as breast, lung and colorectal cancers [31]. SAT1 (Spermidine/spermine N1-acetyltransferase 1) is a rate-limiting enzyme that controls polyamine catabolism in cells [32]. Many stress signals, such as oxidative stress, inflammation and heat shock, activate SAT1 in cells. It has been reported that overexpression of SAT1 leads to the depletion of spermidine and spermine but the increased levels of putrescine, which induces mitochondrial apoptosis and inhibits cell proliferation [33][34]. Recently, SAT1 was shown to be a direct p53 target that can be induced by Nutlin-3 (a small molecule MDM2 inhibitor that activates p53 through disrupting the MDM2-p53 interaction) or the DNA-damaging agent doxorubicin in a p53-dependent manner in cells [35]. ROS-induced cell death in cells with SAT1 overexpression can be inhibited only by ferroptosis inhibitor ferrostatin-1 but not by inhibitors for other types of cell death, such as apoptosis inhibitor Z-VAD-FMK, necroptosis inhibitor necrostatin-1 or autophagy inhibitor 3-methyladenine [35]. SAT1 depletion inhibits p53-regulated ferroptosis in mouse embryonic fibroblasts (MEFs) from both wild-type p53 and p533KR mice [35]. Interestingly, SAT1 does not affect the levels and activities of SLC7A11 and GPX4. In contrast, the levels of ALOX15 (arachidonate 15-lipoxygenase), a member of the lipoxygenase family that oxygenates PUFAs and is responsible for oxidative stress-induced ferroptosis, are elevated upon SAT1 induction. Furthermore, ferroptosis induced by SAT1 and ROS can be effectively blocked by PD146176, an ALOX15-specific inhibitor, indicating that ALOX15 is a mediator of the p53-induced SAT1 expression and ferroptosis [35]. Additionally, ALOX12 (arachidonate 12-lipooxygenase), another member of the lipoxygenase family, was also identified as an important positive regulator for p53-mediated ferroptosis but not for GPX4 and ACSL4-mediated ferroptosis [36]. ALOX12 inactivation abrogates p53-mediated ferroptosis induced by ROS stress and abolishes the tumor suppressive function of p53 in xenograft tumor models. ALOX12 deficiency promotes tumorigenesis in Eμ-Myc lymphoma mouse models. Furthermore, tumor-associated ALOX12 missense mutations lose their lipoxygenase activity and ability to induce ferroptosis [36]. These results suggest that ALOX12 suppresses tumorigenesis through inducing ferroptosis, which contributes to the function of p53 in tumor suppression (Figure 1).
Ferroptosis is also modulated by glutamine metabolism. Upon the deprivation of amino acids, glutamine induces ferroptosis in a serum-dependent manner. In glutamine catabolism, glutamine is firstly converted into glutamate by glutaminases (GLS1 and GLS2) and then further converted into α-ketoglutarate, an important substrate for the citric acid cycle (also named TCA cycle) in mitochondria [37]. GSL2, the liver-type glutaminase in mitochondria, has been identified as a direct transcriptional target of p53; p53 binds to the p53-responsive elements in the GLS2 gene to induce GLS2 transcription [38][39]. The induction of GLS2 mediates the p53 function in oxygen consumption and ATP generation in cells. Moreover, GLS2 promotes cellular antioxidant function by increasing GSH and NADH production in cells [38][39]. GLS2 displays a tumor suppressive function in liver and brain tumors, where GLS2 expression is frequently decreased [38][39][40][41][42]. Interestingly, GLS2 was reported to play a critical role in regulating ferroptosis; knockdown of GLS2 inhibits the serum-dependent ferroptosis induced by deprivation of amino acids. In contrast, GLS1, a kidney-type glutaminase and a homology of GLS2, is not required for ferroptosis. Further studies are necessary to understand whether GLS2 mediates p53-induced ferroptosis and why GLS2 but not GLS1 is required for ferroptosis given they both regulate glutaminolysis in cells.
Cyclooxygenase-2 encoded by the PTGS2 gene is an enzyme that acts both as a peroxidase and a dioxygenase [43]. Cyclooxygenases catalyze lipid oxidation [44][45]. Studies have shown that PTGS2 expression levels are increased during the ferroptosis induced by RSL3, erastin or GPX4-deficiency in mice [46]. Interestingly, PTGS2 expression was reported to be upregulated by ferroptosis in a p53-dependent manner, which was only observed in p533KR/3KRMdm2−/− embryos but not in p53−/−Mdm2−/− embryos. Currently, it is still unclear how p53 regulates PTGS2. Nevertheless, these results suggest that p53 promotes ferroptosis induced by RSL3 and erastin through upregulation of PTGS2 expression.
As mentioned above, iron metabolism is important for ferroptosis. It was reported that p53 regulates iron metabolism through ferredoxin reductase (FDXR), an enzyme that transfers electron from NADPH (nicotinamide adenine dinucleotide phosphate) to cytochrome P450 via ferredoxin in mitochondria [47]. FDXR is a p53 target gene and p53 induces transcriptional expression of FDXR [48]. At the same time, FDXR promotes p53 mRNA translation to increase p53 protein levels through iron regulatory protein 2 in cells [47]. This FDXR-p53 loop has been shown to be critical for tumor suppression through maintaining mitochondrial iron homeostasis; mice heterozygous in FDXR are prone to develop spontaneous tumors and display a short life span compared with wild-type mice [47]. Interestingly, RSL3- and erastin-induced ferroptosis can be suppressed by both FDXR deficiency and FDXR overexpression [47]. Although the underlying mechanism of this observation is unclear, this result suggests an additional potential mechanism of p53 in regulating ferroptosis through modulation of FDXR levels and iron metabolism in cells [47]. Further studies will help to reveal how FDXR and iron metabolism modulate p53-dependent ferroptosis (Figure 1).
In addition, recent studies have also reported that p53 induces ferroptosis through regulating noncoding RNAs. p53 has been reported to regulate expression of many noncoding RNAs, including microRNAs (miRNAs) and long noncoding RNAs (LncRNAs) and meanwhile, p53 is also regulated by many noncoding RNAs [49][50][51][52]. It was reported that p53 binds to the promoter region of RNA-binding protein ELAVL1 (ELAV-like RNA-binding protein 1) to repress the expression of ELAVL1, which binds to and stabilizes LncRNA LINC00336 in lung cancer cells [53]. LINC00336 inhibits ferroptosis by acting as an endogenous sponge of miRNA miR-6852, leading to the enhanced expression of cystathionine-β-synthase (CBS), which is involved in ferroptosis as a marker of transsulfuration pathway activity. Thus, p53 destabilizes LINC00336 and releases miR-6852, which in turn decreases the levels of CBS to promote ferroptosis [53]. Recently, LncRNA PVT1 was reported to induce ferroptosis through inhibition of miR-214-mediated downregulation of transferrin receptor 1 [54]. PVT1 can be induced by p53 and mediate the tumor suppressive function of p53 [55]. Therefore, it is possible that PVT1 mediates the function of p53 in promoting ferroptosis, contributing to the tumor suppressive function of p53. Since miR-214 can also repress p53, p53 may form a positive feedback loop with PVT1 to induce ferroptosis [54]. It was reported that the levels of PVT1 are upregulated and miR-214 levels are downregulated in the plasma from acute ischemic stroke patients [54], which suggests that p53-induced ferroptosis may contribute to ischemic stroke-induced tissue injuries in addition to tumor suppression (Figure 1).
2.2. Wild-type p53 Inhibits Ferroptosis
While many studies using different cultured cell systems and mouse models have shown the promoting effect of p53 on ferroptosis as summarized above, interestingly, some studies also reported that p53 can inhibit ferroptosis. p21 (also known as CDKN1A) was reported to mediate p53 function to delay cystine deprivation-induced ferroptosis in human fibrosarcoma HT-1080 cells [56]. p21 is a well-known p53 target gene that mediates the function of p53 in inducing cell cycle arrest and senescence in response to stress signals . In HT-1080 cells, elevating p53 protein levels by treating cells with the MDM2 inhibitor nutlin-3 attenuates ferroptosis induced by erastin and knockout of p53 by CRISPR/Cas9 technology sensitizes cells to ferroptosis, suggesting a pro-survival role of p53 in ferroptosis [56]. Mechanistically, p53 induces p21 to increase the production of GSH, leading to the reduced accumulation of toxic lipid-ROS to inhibit ferroptosis (Figure 3). p21 exerts its functions in cell cycle arrest through binding to cyclin-dependent kinases (CDKs), including the CDK4/6 complex, and inhibiting their activities [57]. CDK4/6 inhibitors cannot block erastin-induced ferroptosis, which suggests that the inhibition of ferroptosis by p21 is independent of p21-mediated cell cycle arrest [56]. However, in non-cancerous human fetal lung IMR-90 fibroblast cells that express wild-type p53, nutlin-3 treatment increases p53 levels but does not affect the onset of erastin-induced ferroptosis, suggesting that the role of p53 in protection against ferroptosis is not universal [56]. Interestingly, a recent study reported that mutating the CDK binding region of p21 greatly abolishes the inhibitory effect of p21 on ferroptosis, while the PCNA binding-defective mutation of p21 only partially impairs the inhibitory effect of p21 on ferroptosis, which suggests that p21 alters sensitivity to ferroptosis by affecting CDK-mediated functions [58]. Further studies are needed to reveal the mechanism by which p21 regulates ferroptosis.
Recently, p53 was also reported to inhibit ferroptosis in colorectal cancer HCT116 and SW48 cells through binding to DPP4 protein (dipeptidyl peptidase-4; also known as T-cell activation antigen CD26), a multiple functional protease that plays an important role in mediating cell death [59]. The binding of p53 to DDP4 protein regulates the subcellular localization of DPP4 but not the protein levels of DPP4 [59]. In the absence of p53, DPP4 is localized on the plasma membrane, where it forms a complex with NADPH oxidase 1 (NOX1), a member of superoxide-generating NADPH oxidase protein family, to increase lipid peroxidation and ferroptosis. The binding of p53 to DPP4 sequesters DPP4 in a nuclear enzymatic inactive pool, leading to the NOX1 dissociation and decreased lipid peroxidation and ferroptosis [59] (Figure 1). Interestingly, depletion or inhibition of p53 only enhances ferroptosis induced by system xc- inhibitors (e.g., erastin and sulfasalazine) but not ferroptosis induced by GPX4 inhibitors (e.g., RSL3 or FIN56) [59], suggesting that p53 regulates ferroptosis in a ferroptosis inducer-specific manner. Further, DPP4 inhibitors can block erastin-induced ferroptosis in p53-deficient colorectal cancer cells [59]. Interestingly, it was reported that while the expression of DPP4 is low in the normal adult colon, DPP4 is highly expressed in some colon cancers and cancer cell lines [60]. These findings suggest that the function of p53 in inhibiting ferroptosis might be specific for colorectal cancer cells with high DPP4 expression.
Mitochondria have been reported to be involved in different programmed cell death processes, including apoptosis and mitophagy [61][62]. Recently, it was reported that mitochondria are also involved in ferroptosis [63]. Overexpression of Parkin, a protein involved in Parkinson’s disease and mitophagy, was shown to induce the mitophagy and inhibit cysteine deprivation-induced ferroptosis but not GPX4 inhibition-induced ferroptosis in human fibrosarcoma HT-1080 cells [63]. Mechanistically, cysteine deprivation results in the hyperpolarization of mitochondrial membrane potential and production of lipid peroxide, leading to ferroptosis. Blockage of mitochondrial electron transfer chain, TCA cycle or glutaminolysis attenuates mitochondrial membrane potential hyperpolarization, lipid peroxide accumulation and ferroptosis [63]. Interestingly, Parkin is a direct target gene of p53; p53 transcriptionally induce Parkin expression [64][65]. As a p53 target, Parkin mediates the function of p53 in regulating mitochondria respiration, oxygen consumption and anti-oxidant defense [64]. As a tumor suppressor, Parkin has been shown to be involved in metabolic regulation, including suppression of glycolysis and serine synthesis, contributing to the role of p53 in tumor suppression [66][67][68][69]. Currently, it remains unclear whether p53 is involved in cysteine deprivation-induced ferroptosis though regulation of Parkin expression. Taken together, p53 appears to regulate ferroptosis in a highly context-dependent manner, which leads to either positive or negative regulation of ferroptosis (Figure 1).
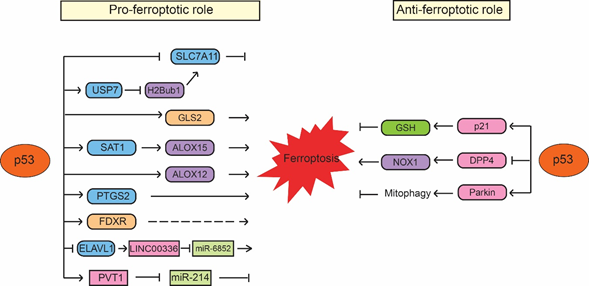
Figure 1. p53 regulates ferroptosis in a context-dependent manner. p53 promotes ferroptosis through its regulation of SLC7A11, GLS2, SAT1/ALOX15, ALOX12, PTGS2, FDXR, as well as the noncoding RNAs, such as LINC00336/miR-6852 and PVT1/miR-214. Meanwhile, p53 also inhibits ferroptosis through its regulation of p21, DPP4 and Parkin. The regulation of ferroptosis by p53 appears to be highly cell/tissue-type and stress signal specific.
2.3. Ferroptosis Regulated by Mutp53, p53 Variants and p53 Family Proteins
In addition to wild-type p53, missense mutp53 has also been reported to be involved in the regulation of ferroptosis. It has been known that NRF2 induces the expression of SLC7A11 to protect cancer cells from ferroptosis [70], whereas wild-type p53 transcriptionally represses SLC7A11 expression to induce ferroptosis. Interestingly, mutp53 was reported to bind to NRF2 and inhibit its transcription activity, reducing the expression of SLC7A11. Tumors with missense mutp53 display decreased levels of SLC7A11 and increased sensitivity to ferroptosis. Further, ectopic SLC7A11 expression in tumors with mutp53 expression promotes resistance of tumors to ferroptosis-inducing drugs (e.g., sulfasalazine), which suggests that mutp53 sensitizes the cancer cells to ferroptosis through repression of SLC7A11 expression. Similar effects of mutp53 were also observed in human colorectal cancer cells; cells expressing mutp53 are more sensitive to erastin-induced ferroptosis than cells expressing wild-type p53 and ectopic expression of R175H mutp53, a GOF mutp53 hotspot mutation, restores the sensitivity to erastin in both HCT116 and SW48 cells. Given that GOF mutp53 often accumulates to very high levels in cancer cells and renders cancer cells resistant to chemotherapy and radiation therapy as a GOF activity, these findings suggest that some cancers expressing GOF mutp53 may be more sensitive to ferroptosis inducers, which can be a potential therapeutic strategy for these cancers. It has been reported that different mutp53 can differ in the magnitudes of their GOF activities and display GOF activities in a cell- or tissue-specific manner [6,123,124]. Therefore, it will be crucial to investigate whether sensitizing cancer cells to ferroptosis inducers is a general phenomenon for different GOF mutp53 in different types of cancers.
Some common single nucleotide polymorphisms (SNPs) in the p53 gene have been known to affect the p53 transcriptional activity and functions in tumor suppression, metabolism, maternal reproduction and aging [71][72][73][74]. The Pro47Ser variant (hereafter S47) is the second most common SNP in the p53 coding region (after Pro72Arg). In a humanized p53 knock-in mouse model of S47 variant, mice expressing homozygous or heterozygous S47 p53 variant are more susceptible to spontaneous cancers of diverse histological types (particularly liver cancer) compared with wild-type mice [75]. Similar to p53-/- MEFs, MEFs with S47 p53 variant display a high GSH/GSSG ratio and low-molecular-weight thiols coenzyme A level and are less sensitive to ferroptosis compared with wild-type MEFs [76]. The S47 p53 variant shows an impaired ability to transcriptionally induce a subset of p53 target genes, including two well-known genes involved in metabolism, GLS2 and SCO2, suggesting that the defect in metabolic regulation may contribute to the reduced ferroptosis and the tumor-prone phenotype observed in S47 mice [75]. Notably, further studies showed that the ferroptotic defect in S47 p53 variant results in iron accumulation in macrophages, which alters macrophage cytokine profiles and leads to the higher level and activity of arginase and decreased activity of nitric oxide synthase [77]. S47 macrophages have decreased liver X receptor (LXR) activation, inflammation and antibacterial defense. S47 mice display more productive intracellular bacterial infections but are protective against malarial toxin hemozoin. Furthermore, iron chelators and LXR agonists can improve the response of S47 mice to bacterial infection [77]. These results suggest that p53-mediated ferroptosis plays an important role in regulation of iron accumulation to modulate macrophage functions and host immune responses [77].
p63 and p73 are two p53 family members, which are homologous to p53 and function as transcription factors like p53 [78][79]. Both p63 and p73 can be transcribed from different promotors resulting in the expression of isoforms with different N-terminal domains, including transcription activation domain (TA) and ΔN (amino-truncated) isoforms. Studies have suggested that the TA isoforms can function as tumor suppressors and induce the expression of canonical p53 target genes, whereas the ΔN isoforms can function as oncogenes and antagonize p53, TAp63 and TAp73 by inhibiting their transcriptional activities [78][79]. Interestingly, ΔNp63α was reported to inhibit both erastin- and RSL3-induced ferroptosis independently of p53 [80]. ΔNp63α transcriptionally regulates GSH biogenesis, utilization and regeneration, leading to the reduction of lipid peroxidation. Furthermore, ΔNp63α upregulates SLC7A11 expression but is unable to inhibit p53-mediated transcriptional repression of SLC7A11. As a result, ΔNp63α induces the expression of SLC7A11 in p53-/- MEFs but not in wild-type MEFs where p53 represses the expression of SLC7A11 [80]. Nevertheless, ΔNp63α protects both wild-type and p53-/- MEFs from ferroptosis induced by erastin and RSL3, indicating that the regulation of GSH homeostasis by ΔNp63α is sufficient to inhibit ferroptosis [80]. Currently, it remains unclear whether p73 is also involved in the regulation of ferroptosis.
2.4. Ferroptosis Regulated by MDM2 and MDM4 Independently of p53
Although MDM2 and MDM4 are critical negative regulators of p53, many studies have shown that MDM2 and MDM4 also exert oncogenic effects independently of their activities to inhibit p53 function. Interestingly, a recent study reported that MDM2 and MDM4 also display a p53-independent function to promote ferroptosis [81]. Both inhibitors of MDM2 (MEL23) and MDM4 (NCS207895) protect cells from ferroptosis induced by erastin and RSL3 through enhancing the levels of ferroptosis suppressor protein 1 (FSP1), an enzyme that reduces coenzyme Q10, an endogenous lipophilic antioxidant, to ubiquinol, which in turn enhances the levels of coenzyme Q10 [81]. Further, the MDM2-MDM4 complex reprograms lipid metabolism by altering the activity of PPARα, leading to enhanced lipid peroxidation and ferroptosis independently of p53 [81]. These results suggest that cancers with MDM2/MDM4 amplification and/or overexpression might be more sensitive to ferroptosis inducers, which can be tested as a potential therapeutic strategy for these cancers.
2.5. p53 Regulation in Response to Ferroptosis Stimuli and Inhibition
As a stress sensor, p53 responds to many different types of stress signals and initiate its transcription program to regulate different stress responses . While many studies have shown that p53 is involved in the regulation of ferroptosis in a highly context-dependent manner, an interesting question raised is how p53 responds to different ferroptosis inducers and inhibitors to regulate ferroptosis. Some studies have shown that p53 can be activated by ferroptosis inducers or inhibitors. For example, it was reported that erastin can induce p53 protein levels and increase p53 transcriptional activity towards its target genes, including p21 and Bax, in human lung cancer A549 cells [134]. Treating cells with ROS scavenger N-acetyl-1-cysteine (NAC) abolishes the effect of erastin on p53 levels, which suggests that enhancing the ROS levels is a mechanism contributing to p53 activation by erastin [82]. Compound D13, a derivative of the triterpene saponin natural compound Albiziabioside A, was reported to induce ferroptosis through suppressing GPX4 expression in human HCT116 colorectal cancer cells [83]. D13 displays an anti-tumor effect in xenograft tumors formed by HCT116 cells. Furthermore, D13 activates p53 and displays a p53-dependent effect on ferroptosis [83]. Interestingly, iron chelators, including desferrioxamine, have been reported to activate p53 [84][85][86]. HIF-1α may play an important role in mediating the induction of p53 by iron chelating agents. It has been reported that iron deprivation caused by iron chelating agents results in the activation of HIF1α [87][88] and HIF-1α stabilizes p53 through inhibiting the ubiquitination and nuclear export of p53 [89][90]. A recent study reported that the iron polyporphyrin heme inhibits p53 function by binding to p53 and leading to nuclear export and degradation of p53 [91]. Therefore, iron deprivation induced by desferrioxamine activates p53 and suppresses growth of HCT116 xenograft tumors in a p53-dependent manner. However, it is unclear how ferroptosis contributes to this the tumor suppressive effect of desferrioxamine and how p53 activation by desferrioxamine contributes to ferroptosis in this study [91]. Ferroptosis inhibitors, including ferrostatins, liproxstatins and iron chelators, have been reported to display protective effects on ischemic injuries and neurodegenerative diseases . The iron chelator deferiprone was reported to display a beneficial effect on Parkinson’s disease patients . Interestingly, p53 has been reported to be involved in ischemic injuries and neurodegenerative diseases . Currently, it remains largely unknown how p53 responds to these ferroptosis inhibitors and how p53 is involved in ferroptosis in these conditions and models. Given the critical roles of p53 and ferroptosis in the development and therapy of human diseases, including cancer, ischemic injuries and neurodegenerative diseases, future studies are needed to further elucidate how p53 responds to different ferroptosis inducers and inhibitors to regulate ferroptosis in a cell-, tissue-type and disease-specific manner.
References
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292.
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530.
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, doi:10.1093/jmcb/mjaa040. Online ahead of print.
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286.
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317.
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606.
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5.
- Ho, T.; Tan, B.X.; Lane, D. How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. Int. J. Mol. Sci. 2019, 21, 13.
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell Oncol. 2014, 1, e955995.
- Mantovani, F.; Collavin, L.; del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212.
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2020, 1–28. doi: 10.1080/15548627.2020.1810918. Online ahead of print.
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414.
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285.
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang. G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88.
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296.
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245.
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2018, 40, 382–395.
- Wang, S.J.; Ou, Y.; Jiang, L.; Gu, W. Ferroptosis: A missing puzzle piece in the p53 blueprint? Mol. Cell Oncol. 2016, 3, e1046581.
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front. Endocrinol. 2018, 9, 124.
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168.
- Jiang, L.; Kon, N.; Li, T.; Wang, S. J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62.
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2020, 1–22. doi: 10.1007/s13238-020-00789-5. Online ahead of print.
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadee, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454.
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373.
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q. R.; He, Q.; Zhao, S.; Zhang, G. A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563.
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792.
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390.
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E995–E1010.
- Mandal, S.; Mandal, A.; Johansson, H. E.; Orjalo, A. V.; Park, M. H. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2169–2174.
- Mandal, S.; Mandal, A.; Park, M.H. Depletion of the polyamines spermidine and spermine by overexpression of spermidine/spermine N(1)-acetyltransferase 1 (SAT1) leads to mitochondria-mediated apoptosis in mammalian cells. Biochem. J. 2015, 468, 435–447.
- Ou, Y.; Wang, S. J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812.
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591.
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–34.
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460.
- Suzuki, S.; Tanaka, T.; Poyurovsky, M. V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y. et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466.
- Szeliga, M.; Bogacinska-Karas, M.; Rozycka, A.; Hilgier, W.; Marquez, J.; Albrecht, J. Silencing of GLS and overexpression of GLS2 genes cooperate in decreasing the proliferation and viability of glioblastoma cells. Tumour Biol. 2014, 35, 1855–1862.
- Martin-Rufian, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A. R.; Campos-Sandoval, J. A.; Gomez-Garcia, M. C.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J. A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. 2014, 92, 277–290.
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K. H.; Hu, W.; Feng, Z. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 2014, 5, 2635–2647.
- Amadio, P.; Tarantino, E.; Sandrini, L.; Tremoli, E.; Barbieri, S. S. Prostaglandin-endoperoxide synthase-2 deletion affects the natural trafficking of Annexin A2 in monocytes and favours venous thrombosis in mice. Thromb. Haemost. 2017, 117, 1486–1497.
- Bretscher, P.; Egger, J.; Shamshiev, A.; Trotzmuller, M.; Kofeler, H.; Carreira, E. M.; Kopf, M.; Freigang, S. Phospholipid oxidation generates potent anti-inflammatory lipid mediators that mimic structurally related pro-resolving eicosanoids by activating Nrf2. EMBO Mol. Med. 2015, 7, 593–607.
- Liu, X.; Moon, S. H.; Jenkins, C. M.; Sims, H. F.; Gross, R. W. Cyclooxygenase-2 Mediated Oxidation of 2-Arachidonoyl-Lysophospholipids Identifies Unknown Lipid Signaling Pathways. Cell Chem. Biol. 2016, 23, 1217–1227.
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103.
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y. S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256.
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T. A.; Murphy, M. P.; Kelso, G. F.; Smith, R. A.; Kinzler, K. W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117.
- Dangelmaier, E.; Lazar, S.B.; Lal, A. Long noncoding RNAs: p53’s secret weapon in the fight against cancer? PLoS Biol. 2019, 17, e3000143.
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of p53. J. Cell Biochem. 2017, 118, 7–14.
- Chaudhary, R.; Lal, A. Long noncoding RNAs in the p53 network. Wiley Interdiscip. Rev RNA 2017, 8, e1410.
- Luo, Z.; Cui, R.; Tili, E.; Croce, C. Friend or Foe: MicroRNAs in the p53 network. Cancer Lett. 2018, 419, 96–102.
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343.
- Lu, J.; Xu, F.; Lu, H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020, 260, 118305.
- Olivero, C.E.; Martinez-Terroba, E.; Zimmer, J.; Liao, C.; Tesfaye, E.; Hooshdaran, N.; Schofield, J. A.; Bendor, J.; Fang, D.; Simon, M. D.; et al. p53 Activates the Long Noncoding RNA Pvt1b to Inhibit Myc and Suppress Tumorigenesis. Mol. Cell 2020, 77, 761–774.e8.
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K. T.; Li, Y.; Ye, J.; Attardi, L. D.; Dixon, S. J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575.
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414.
- Venkatesh, D.; Stockwell, B.R.; Prives, C. p21 can be a barrier to ferroptosis independent of p53. Aging 2020, 12, 17800–17814.
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T. R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704.
- Havre, P.A.; Abe, M.; Urasaki, Y.; Ohnuma, K.; Morimoto, C.; Dang, N. H. The role of CD26/dipeptidyl peptidase IV in cancer. Front Biosci. 2008, 13, 1634–1645.
- Praharaj, P.P.; Naik, P. P.; Panigrahi, D. P.; Bhol, C. S.; Mahapatra, K. K.; Patra, S.; Sethi, G.; Bhutia, S. K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: Its implication in cancer therapeutics. Cell Mol. Life Sci. 2019, 76, 1641–1652.
- Hou, X.S.; Wang, H. S.; Mugaka, B. P.; Yang, G. J.; Ding, Y. Mitochondria: Promising organelle targets for cancer diagnosis and treatment. Biomater. Sci. 2018, 6, 2786–2797.
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A. M.; Monian, P.; Thompson, C. B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3.
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A. J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264.
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J. Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–75.
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C. A.; et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823.
- Liu, J.; Zhang, C.; Wu, H.; Sun, X. X.; Li, Y.; Huang, S.; Yue, X.; Lu, S. E.; Shen, Z.; Su, X.; et al. Parkin ubiquitinates phosphoglycerate dehydrogenase to suppress serine synthesis and tumor progression. J. Clin. Invest. 2020, 130, 3253–3269.
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. 2018, 38, 40.
- Gupta, A.; Anjomani-Virmouni, S.; Koundouros, N.; Dimitriadi, M.; Choo-Wing, R.; Valle, A.; Zheng, Y.; Chiu, Y. H.; Agnihotri, S.; Zadeh, G.; et al. PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol. Cell 2017, 65, 999–1013.e7.
- Fan, Z.; Wirth, A. K.; Chen, D.; Wruck, C. J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371.
- Mello, S.S.; Attardi, L.D. Not all p53 gain-of-function mutants are created equal. Cell Death Differ. 2013, 20, 855–857.
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S. X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909.
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C. P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A. V.; Murphy, M. E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev. 2018, 32, 230–243.
- Barnoud, T.; Parris, J.L.D.; Murphy, M.E. Common genetic variants in the TP53 pathway and their impact on cancer. J. Mol. Cell Biol. 2019, 11, 578–585.
- Jennis, M.; Kung, C. P.; Basu, S.; Budina-Kolomets, A.; Leu, J. I.; Khaku, S.; Scott, J. P.; Cai, K. Q.; Campbell, M. R.; Porter, D. K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930.
- Leu, J.I.; Murphy, M.E.; George, D.L. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl. Acad. Sci. USA 2019, 116, 8390–8396.
- Singh, K.S.; Leu, J. I.; Barnoud, T.; Vonteddu, P.; Gnanapradeepan, K.; Lin, C.; Liu, Q.; Barton, J. C.; Kossenkov, A. V.; George, D. L.; et al. African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nat. Commun. 2020, 11, 473.
- Dotsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. p63 and p73, the ancestors of p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a004887.
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A. J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198.
- Wang, G.X.; Tu, H. C.; Dong, Y.; Skanderup, A. J.; Wang, Y.; Takeda, S.; Ganesan, Y. T.; Han, S.; Liu, H.; Hsieh, J. J.; et al. DeltaNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep. 2017, 21, 2926–2939.
- Venkatesh, D.; O'Brien, N. A.; Zandkarimi, F.; Tong, D. R.; Stokes, M. E.; Dunn, D. E.; Kengmana, E. S.; Aron, A. T.; Klein, A. M.; Csuka, J. M.; et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020, 34, 526–543.
- Huang, C.; Yang, M.; Deng, J.; Li, P.; Su, W.; Jiang, R. Upregulation and activation of p53 by erastininduced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol. Rep. 2018, 40, 2363–2370.
- Wei, G.; Sun, J.; Hou, Z.; Luan, W.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Novel antitumor compound optimized from natural saponin Albiziabioside A induced caspase-dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur. J. Med. Chem. 2018, 157, 759–772.
- Saletta, F.; Suryo Rahmanto, Y.; Noulsri, E.; Richardson, D. R. Iron chelator-mediated alterations in gene expression: Identification of novel iron-regulated molecules that are molecular targets of hypoxia-inducible factor-1 α and p53. Mol. Pharmacol. 2010, 77, 443–458.
- Kim, B.M.; Choi, J. Y.; Kim, Y. J.; Woo, H. D.; Chung, H. W. Desferrioxamine (DFX) has genotoxic effects on cultured human lymphocytes and induces the p53-mediated damage response. Toxicology 2007, 229, 226–235.
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S. V.; Torti, F. M. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918.
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32.
- Peyssonnaux, C.; Zinkernagel, A. S.; Schuepbach, R. A.; Rankin, E.; Vaulont, S.; Haase, V. H.; Nizet, V.; Johnson, R. S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest. 2007, 117, 1926–1932.
- An, W.G.; Kanekal, M.; Simon, M. C.; Maltepe, E.; Blagosklonny, M. V.; Neckers, L. M. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 1998, 392, 405–408.
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J. Biol. Chem. 2003, 278, 13595–13598.
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193.

