 +1 credit
+1 credit
Video Upload Options
The relationship between periodontitis and systemic diseases, notably including atherosclerosis and diabetes, has been studied for several years. Porphyromonas gingivalis, a prominent component of oral microorganism communities, is the main pathogen that causes periodontitis. We comprehensively summarize the adverse effects of Porphyromonas gingivalis on multiple systems and a variety of diseases, from extensively studied fields (cardiovascular diseases, cancer, adverse pregnancy outcomes, etc.) to emerging areas (Alzheimer's Disease, nonalcoholic fatty liver disease, depression, etc.). Although a few results remain controversial, it is now evident that Porphyromonas gingivalis should be regarded as a modifiable factor for several diseases.
1. Characteristics of Porphyromonas gingivalis (P. gingivalis)
P. gingivalis, one of over 700 bacterial species in the oral cavity, is a Gram-negative, anaerobic, rod-shaped bacteria that forms black colonies on blood agar and requires the presence of heme or hemin and vitamin K in its growth milieu. It is a successful colonizer of the oral epitheliumand an important component of subgingival microbiomes[1]. P. gingivalisis responsible for the chronic form of periodontitis, as it can remodel the commensal bacterial community to promote a state of dysbiosis[2]. Throughout evolution, it has developed unique and intricate mechanisms, such as the alteration of signaling pathways of inflammation, the complement system, the cell cycle, and apoptosis, and the interaction with various host receptors, thereby engineering its environment or modifying the host’s immune response to modulate the entire ecosystem and to persist in host tissues[3]. The survival strategies and pathogenicity of P. gingivalis largely depend on its diverse virulence factors, including its own structural components (lipopolysaccharide, fimbriae, heat shock proteins, etc.) and secretory components (gingipains and outer membrane vesicles).
Fimbriae are crucial for enabling P. gingivalis to specifically bind to eukaryotic cells and other species of bacteria to enhance bacterial motility, biofilm formation, and bacterial invasion of the cells [4]. It can also activate various host cells and subvert host immune clearance[4]. P. gingivalis lipopolysaccharide (LPS) can trigger the innate immune response via activation of Toll-like receptors (TLRs)[5]. The lipid A component of P. gingivalis LPS exhibits two predominant variations of acylation that are attributed to different strains and microenvironmental conditions: the penta-acylated LPS activates TLR4, while tetra-acylated LPS acts as a TLR4 antagonist and TLR2 agonist[5]. The heat shock protein 60 (HSP60) component of P. gingivalis is remarkably immunogenic and plays a critical role in P. gingivalis-induced autoimmune diseases[6]. Gingipains, which consist of lysine-gingipain (Kgp) and arginine-gingipain (Rgp), have multiple impacts on both innate and acquired immunity. These enzymes play essential roles in host colonization, host defense deactivation, tissue destruction, and nutrient acquisition[7]. Outer membrane vesicles (OMVs) from P. gingivalis are enriched in major virulence mediators, such as gingipains, LPS, and the capsule, and participate in biofilm development, host interaction, colonization, and immune defense evasion[8]. Moreover, its characteristic features, such as concentrated gingipains, together with its ability to travel to distant sites, might participate in P. gingivalis-associated systemic disorders[9].
P. gingivalis in local periodontal tissue can enter the vasculature through ulcerated epithelium[10] and lymph vessels[11] shortly after everyday activities, such as brushing and chewing, along with dental procedures. Some studies have indicated that P. gingivalis can survive in other organs besides the oral cavity. Viable P. gingivalis has been detected in human atherosclerotic plaque tissues[12] and mouse lungs[13] through tissue homogenates that were incubated with cells or cultured directly on blood agar plates. Cellular experiments have also provided evidence for the survival of P. gingivalis in some cells. For example, live P. gingivalis has been isolated from human aortic endothelial cells[14], human pancreatic tumor cells[15], and human myeloid dendritic cells[16]. All of the properties described above confer this species with the ability to invade distant tissues, where it is then involved in the onset and/or progression of systemic diseases.
2. P. gingivalis and systemic diseases
Chronic periodontitis, a multifactorial chronic inflammatory disease resulting from dysbacteriosis, is characterized by the destruction of connective tissue and alveolar bone, and it has become the primary reason for tooth loss in adults. It affects nearly 50% of the population worldwide, representing one of the most common inflammatory diseases in humans[17]. Over the past two decades, mounting evidence has supported periodontitis as a potential risk factor for multiple systemic diseases, for example, cardiovascular diseases. As the key etiological agent in periodontitis, Porphyromonas gingivalis (P. gingivalis) has proved to be closely correlated with the occurrence and development of many systemic diseases, such as atherosclerosis, cancer, and Alzheimer’s disease[18][19][20]. The roles of P. gingivalis in systemic diseases have been discussed for several years. In this review, we systematically and comprehensively provide an update and summary of the literature on P. gingivalis-related systemic diseases that affect the whole body, as well as the internal mechanisms, to provide a more comprehensive understanding of P. gingivalis and its relationship with systemic diseases.
2.1 Atherosclerotic cardiovascular diseases (ACVDs)
ACVDs, including coronary artery disease and stroke, are a form of CVDs with high morbidity and mortality rates. Atherosclerosis (AS), resulting from the progressive accumulation of lipids, calcium, macrophages, and other components in the artery wall, is the pathological basis of ACVDs. Trials in humans have found P. gingivalis in clinical samples at a detection rate of 82.61% by fluorescent in situ hybridization assay[21]. Subsequently, in vivo experiments confirmed the promoting effect of P. gingivalis on AS. Infection with P. gingivalis exacerbated atherogenesis in apolipoprotein E (ApoE)-deficient mice[18][21], and the proximal aortic lesion size in P. gingivalis-inoculated mice was 2-fold larger than that in control mice[18].
Most scholars regard AS as an excessive inflammatory response after arterial wall endothelial dysfunction resulting from many damage factors. The pathogenesis mechanisms of AS are highly complex and include the activation of endothelial cells and platelets, recruitment of leukocytes (mainly monocytes and macrophages), migration and proliferation of smooth muscle cells (SMCs), and formation of a lipid core, along with thrombosis and plaque instability. Studies have shown that P. gingivalis can promote AS by affecting the function of all of these cells . First, P. gingivalis can activate endothelial cells and induce endothelial dysfunction. As reported in a previous review, P. gingivalis invades endothelial cells via the autophagic pathway while suppressing apoptosis [22]. Through the NF-κB or p38 MAPK pathway, its fimbria and LPS positively upregulate the expression of various adhesion molecules in endothelial cells, such as vascular cell adhesion molecule-1, intercellular adhesion molecule-1, monocyte chemoattractant protein, P-selectin, and E-selectin[23][24]. This is an essential step in the pathogenesis of endothelial dysfunction. In recently published research, it was revealed that P. gingivalis enhanced oxidative stress and the inflammatory response in aortic endothelial cells via the NF-κB-BMAL1-NF-κB signaling loop, leading to the aggravation of AS[21] . Furthermore, P. gingivalis can induce procoagulant effects in endothelial cells, and this prothrombotic response may be associated with plaque progression and instability[25]. Second, it has been reported that IgG-opsonized P. gingivalis may bind to the FcγRIIa receptor on platelets and activate GPIIb⁄IIIa integrin, which becomes connected to Hgp44 adhesin through a fibrinogen bridge, inducing further platelet activation and aggregation[26]. A recent review analyzed the interactions of P. gingivalis with activated platelets, and the overall outcome may be the modified expression of CKs, which might affect the inflammatory response and fibrinolysis[27]. Third, P. gingivalis and its virulence components (such as LPS, fimbria) are involved in each phase of monocyte activity during the formation of AS by supporting monocyte migration to the endothelial surface, intimal infiltration, and differentiation into pro-inflammatory macrophages and eventually foam cells[28][29]. Fourth, P. gingivalis can play a distinct role in foam cell formation, which is a critical step in the atherosclerotic process. P. gingivalis LPS promotes the accumulation of lipids in macrophages and the formation of macrophage-derived foam cells by upregulating CD36 (a scavenger receptor for low-density lipoprotein and oxidized low-density lipoprotein) as a result of c-Jun/AP-1 pathway activation and by downregulating ATP-binding cassette transporter A1 (cholesterol efflux moderator) due to increased calpain activity[29]. A study in 2019 experimentally determined that P. gingivalis promoted lipid uptake in macrophages by inducing the expression of fatty acid-binding protein 4, which may be dependent on the JNK pathway[30]. In addition, P. gingivalis enhanced the TLR2-CD36/SR-B2-dependent systemic release of IL-1β, leading to a subsequent increase in lipid uptake by macrophages and foam cell formation as a result of encountering IL-1β in the vessel wall[31]. Furthermore, the proteolytic activity of Rgp and Kgp was shown to induce lipid peroxidation and to change the expression of low-density lipoprotein and high-density lipoprotein, causing foam cell formation from macrophages[32]. Evidence from these studies suggests that lipoproteins play a key role in the connection between P. gingivalis and AS progression. P. gingivalis infection may also contribute to AS by modulating lipid metabolism and homeostasis. Fifth, vascular calcification is a prominent feature of AS. P. gingivalis LPS can stimulate the proliferation and calcification of SMCs, resulting in vascular calcification[33]. P. gingivalis OMVs were also reported to promote vascular SMC calcification through ERK1/2-RUNX2[34]. Additionally, WADA et al. proposed a putative molecular mechanism in which P. gingivalis could contact or invade the SMC layer in blood vessels after endothelial cell injury and induce the expression of S100A9 (a member of the S100 calcium-binding protein family), which triggers the transformation of SMCs from a contractile to a proliferative phenotype, stimulating further cell growth and contributing to aortic intimal hyperplasia[35].
2.2 Oral squamous cell carcinoma (OSCC)
OSCC often occurs in the tongue, floor of the mouth, buccal mucosa, and gingiva. A meta-analysis revealed that periodontitis increased the risk of OSCC by nearly 2-fold[36]. Furthermore, P. gingivalis was shown to increase the chance of OSCC, and its colonization in tumor tissues has reduced patient survival[37]. The detection of P. gingivalis by immunohistochemical staining was more than 33% higher in gingival carcinoma tissues than in normal gingival tissues[38]. Afterwards, in vivo experiments further confirmed the negative effect of P. gingivalis in OSCC. It was found that P. gingivalis accelerated OSCC progression in an immune microenvironment through the secretion of CCL2, CXCL2, IL-6, and IL-8 from infected oral dysplastic keratinocytes to recruit myeloid-derived suppressor cells[37]. In addition, oral administration of P. gingivalis promoted 4-nitroquinoline-1-oxide-induced tongue tumorigenesis and aggravated the disturbance of fatty acid metabolism during oral carcinoma progression[19].
Large studies have explored the mechanisms of P. gingivalis in OSCC, including epithelial–mesenchymal transition (EMT) of oral epithelial cells, the inhibition of epithelial cell apoptosis, the promotion of immune evasion, the proliferation and invasion of tumor cells, and so on . First, P. gingivalis upregulates the levels of zinc-finger E-box-binding homeobox proteins (ZEB1 and ZEB2), which are transcription factors that regulate EMT through GSK-3β[39] and β-catenin/forkhead box-O1 (FOXO1), respectively[40]. A review by Olsen provided a summary of evidence related to P. gingivalis-induced EMT of OSCC cells and human primary oral epithelial cells[41]. Second, P. gingivalis modulates epithelial cell apoptosis via multiple anti-apoptotic/survival pathways, including the activation of the PI3K/Akt and JAK/Stat pathways, release of survivin, upregulation of anti-apoptotic Bad and Bcl-2, downregulation of pro-apoptotic Bax, and inhibition of cytochrome c release and caspase-9 and caspase-3 activation[42][43]. P. gingivalis infection upregulates miR-203, which directly inhibits suppressor of cytokine signaling 3 and leads to increased Stat3 activation[44], which may also be involved in the anti-apoptotic mechanism of the epithelium, given the role of Jak/Stat mentioned above. In addition, nucleoside diphosphate kinase (NDK), an ecto-ATPase secreted by intracellular P. gingivalis, confers epithelial cells with an anti-apoptotic phenotype by binding to and phosphorylating HSP27, which inhibits cytochrome c release and caspase-9 activation[45]. NDK from P. gingivalis can also scavenge ATP and inhibit P2X7-mediated host-cell apoptosis[46]. Moreover, P. gingivalis infection promotes a prosurvival phenotype in human primary oral epithelial cells by regulating cyclins and p53[47]. P. gingivalis-induced ROS activates the multipurpose transcriptional regulator FOXO1 via JNK signaling and then initiates an anti-apoptotic program in epithelial cells[48]. Third, internalized P. gingivalis upregulates the expression of B7-H1 and B7-DC receptors on oral cancer cells via a receptor-interacting serine/threonine-protein kinase 2-dependent mechanism, contributing to the escape of tumor cells from immunosurveillance[49]. Furthermore, NDK from P. gingivalis has antagonist effects on ATP activation of P2X7 receptors, leading to reduced IL-1β production in epithelial cells, thereby promoting the immune evasion of tumor cells[50]. Fourth, P. gingivalis promotes the proliferation of OSCC cells by regulating the expression of cyclin D1 through the miR-21/PDCD4/AP-1 negative signaling pathway[51]. Fifth, P. gingivalis-infected OSCC cells can increase invasiveness through EMT-like changes[52]. In addition to EMT, P. gingivalis may have the ability to induce proMMP9 expression via ERK1/2-Ets1, p38/HSP27, and PAR2/NF-kB pathways, which subsequently activate MMP9, promoting OSCC cell invasion[53]. P. gingivalis exposure also increases the invasive ability of oral cancer cells via the upregulation of MMPs in an IL-8-dependent fashion, including MMP-1, MMP-2, and MMP-10[52][54]. Beyond the mechanisms mentioned above, it has been reported that inflammatory mediators elicited by P. gingivalis could induce cell proliferation, mutagenesis, oncogene activation, angiogenesis, and immunosuppression, thus facilitating the development of OSCC[55].
2.3 Alzheimer's Disease (AD)
AD is a neurodegenerative disease. It is the most common reason for dementia and is becoming a major health problem in aging societies worldwide. A historical cohort study found that chronic periodontitis patients had an elevated risk of AD[56], and P. gingivalis DNA, LPS[57], and gingipains[58] have been identified in AD brains. Parallel epidemiological studies and animal experiments also strengthen support for the possible relevance of P. gingivalis in AD pathogenesis. P. gingivalis exposure induced AD-like phenotypes in mice, which presented as microglia-mediated neuroinflammation, β-amyloid (Aβ) accumulation in neurons, impaired cognitive function, and a reduction in learning and memory[59][60].
The pathology of AD has three major hallmarks: Aβ plaques, neurofibrillary tangles, and microglia-mediated neuroinflammation. P. gingivalis may be involved in the progression of AD through these processes. Cathepsin B (CatB) is crucial for Aβ deposition and the mediation of neuroinflammation. Intraperitoneal injection of P. gingivalis LPS into middle-aged WT mice significantly increased CatB expression in both microglia and neurons, and P. gingivalis LPS-induced AD-like phenotypes were found to occur only in a CatB-dependent manner[59]. Furthermore, in P. gingivalis-infected mice, Aβ accumulated in inflammatory monocytes/macrophages, mainly based on CatB/NF-κB signaling activation, and this is the first evidence to suggest that inflammatory monocytes/macrophages act as the source of peripheral Aβ when exposed to P. gingivalis[61]. Therefore, considering the pivotal role of CatB, it has been regarded as a potential therapeutic target for preventing P. gingivalis-associated cognitive decline in AD[59][61]. In addition, hippocampal neurons had significantly increased mean mRNA levels of amyloid precursor protein and CatB when treated with conditioned medium from P. gingivalis LPS-treated WT primary microglia, but not when treated with P. gingivalis LPS directly[59]. Accordingly, the pivotal, detrimental role of P. gingivalis against microglia is involved in the pathogenesis and development of AD. A recent review indicated that P. gingivalis may affect microglia and genes such as apolipoprotein, clusterin, CD33, and complement receptors to cooperatively promote the neurodegeneration that is typical of AD[62]. Furthermore, P. gingivalis LPS might induce neuroinflammation and cognitive impairment in mice via the TLR4/NF-κB signaling pathway[60]. In addition to the direct role of P. gingivalis, the release of inflammatory molecules may also be involved. Various host cells infected with P. gingivalis reportedly release a set of CKs, such as TNF-α, IL-1, IL-6, and IL-8, in their immune response[63]. Pro-inflammatory mediators could reach the central nervous system via hematoencephalic barrier-free areas and fenestrated capillaries or by regulating blood–brain barrier permeability[64]. Systemic inflammation, including inflammatory mediators such as CRP, TNF-a, IL-6, and IL-1β, may also increase the risk and progression of cognitive decline and AD. In contrast to the above-described studies, Liu et al. focused on gingipains, Rgp and Kgp. They revealed that gingipains cooperatively contributed to the cell migration of microglia towards the infected site and induced neuroinflammation through the proteolytic activation of PAR2, and the subsequent activation of PI3K/Akt and MEK/ERK pathways may also play fundamental roles in this process[65]. In addition, researchers have proposed that P. gingivalis may also be involved in the progress of AD by contributing to the suppression of the host's adaptive immune system. P. gingivalis could prevent the entry of immune cells into the brain, increase blood–brain barrier permeability, and inhibit the local IFN-γ response. The scarcity of adaptive immune cells in AD neuropathology further implies that P. gingivalis residing in the brain may impair clearance of Aβ and induce immunosuppression[66].
2.4 Diabetes
Diabetes, a series of chronic metabolic disorders characterized by hyperglycemia, is the result of defects in insulin secretion, insulin action, or both. Considerable epidemiologic evidence has shown a two-way interrelationship between periodontitis and diabetes, and many researchers regard periodontal infection as a risk factor for diabetes progression. Diabetic individuals with periodontitis were found to have a significantly higher prevalence of diabetes-related complications[67].Further animal experiments have confirmed that P. gingivalis or its components can induce insulin resistance in mice[68][69][70][71], which is one of the vital pathogeneses of type 2 diabetes.
Among the potential P. gingivalis-related mechanisms involved in the course of diabetes, insulin resistance is considered to be the most important. P. gingivalis infection increased systemic inflammation, especially in adipose tissue, through the induction of endotoxemia [69], alteration of gut microbiota[72], or an impaired regional adaptive immune response [68], ultimately resulting in insulin resistance. Moreover, inflammation and elevated levels of inflammatory markers, such as CRP and IL-6, can induce insulin resistance[73]. Thus, increased levels of systemic pro-inflammatory CKs initiated by P. gingivalis may promote insulin resistance. P. gingivalis LPS induced pro-inflammatory adipokine secretion and oxidative stress in adipocytes through the regulation of TLR-mediated pathways and redox enzymes and contributed to obesity-associated insulin resistance[74]. A recent study in 2020 indicated that P. gingivalis aggravated high-fat diet (HFD)-induced insulin resistance in mice via its biosynthesis of branched-chain amino acids, which can activate the mammalian target of rapamycin and downstream genes, which leads to the dephosphorylation of insulin receptor substrate 1[71]. Furthermore, P. gingivalis OMVs were loaded with gingipains and translocated to the liver in mice, leading to the attenuation of insulin sensitivity and inhibition of hepatic glycogen synthesis, partly by attenuating the Akt and GSK-3β pathway[70]. Importantly, potential mechanisms other than insulin resistance should be examined. Higher colonization levels of P. gingivalis are reported to be linked to higher prediabetes prevalence among diabetes-free adults[75]. P. gingivalis LPS stimulated insulin secretion by pancreatic β-cells and had significant implications in the progression of β-cell compensation in prediabetes in subjects with periodontitis[76]. Moreover, oral administration of P. gingivalis induced the translocation of P. gingivalis/gingipains to the pancreas in mice, leading to significant changes in islet architecture and β-cell apoptosis, which may be involved in the development of prediabetes[77]. Lastly, P. gingivalis attenuated the phosphorylation and translocation of FOXO1, which is regulated by insulin in HepG2 cells, thereby increasing hepatic gluconeogenesis[78] and potentially leading to elevated blood glucose.
3. Conclusion
The latest studies have tended to focus on exploring possible direct and indirect links between P. gingivalis and certain systemic diseases, especially AD and AS, and an increasing amount of evidence has been accumulating. P. gingivalis invades the human body and influences general health in four main ways (Figure 1). (i) Bacteremia: The direct invasion of P. gingivalis in epithelium, endothelial cells, and subepithelial tissues has been demonstrated[79]. Transient bacteremia of P. gingivalis frequently occurs during daily activities, such as toothbrushing, chewing, and flossing, and after dental treatment procedures[10], which enable the bacteria to enter the rest of the body and take part in local pathogenesis. (ii) Immunologic sounding: Persistent local infection caused by P. gingivalis induces the upregulation of inflammatory cascades involving IL-1, IL-6, IL-8, TNF-α, CRP, and IFN[63]. A low-level, long-term systemic inflammatory status might be implicated in the pathology of systemic disorders. (iii) Specific toxins of P. gingivalis, such as gingipains and OMVs, have been detected at multiple body sites (such as the brain and liver) and play pivotal roles in the progression of local diseases[58][70]. (iiii) Pathogen trafficking: Direct infection and internalization in host immune cells, such monocytes, and subsequent recruitment to tissues throughout the whole body may be another pathogenic strategy of P. gingivalis[58].
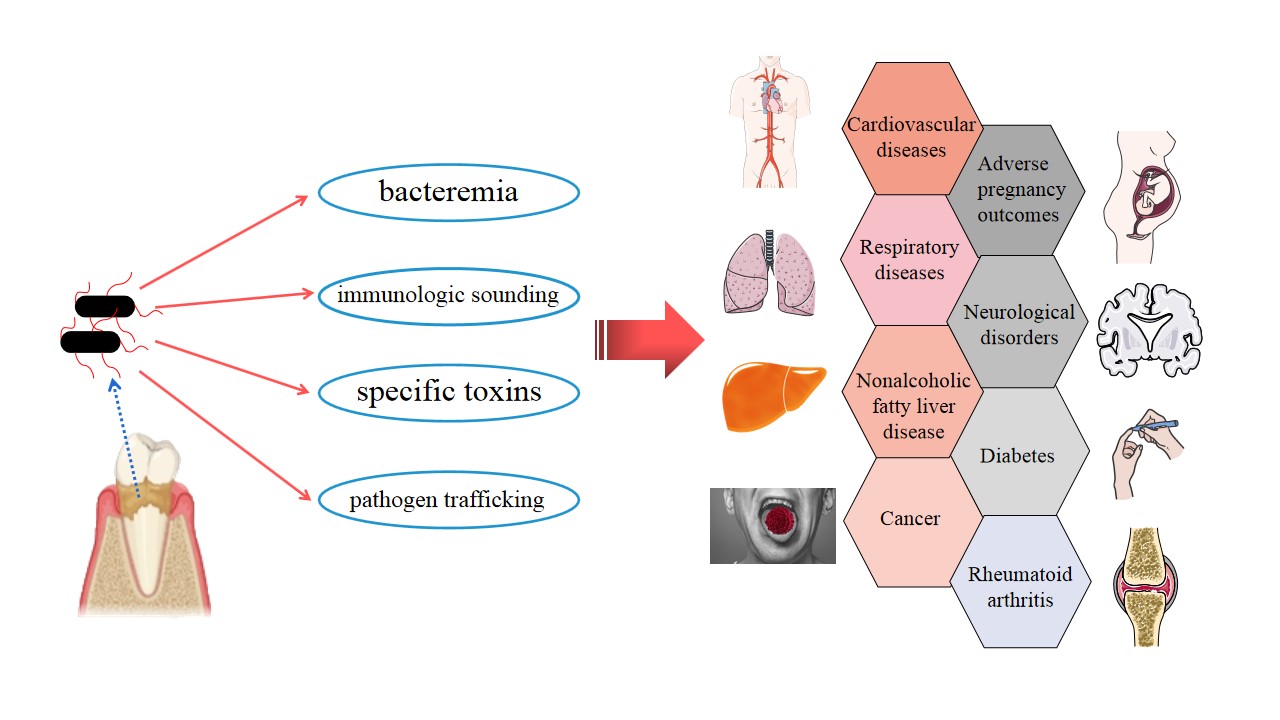
Figure 1. Strategies by which Porphyromonas gingivalis can invade the whole body, along with simple a schematic representation of Porphyromonas gingivalis-associated systemic diseases.
In this article, we comprehensively summarize the adverse effects of P. gingivalis on multiple systems and a variety of diseases. Some of the clearer and more direct mechanisms are discussed here. However, many of the exact mechanisms are not fully clear yet, and a few conclusions are controversial. Current evidence emphasizes that promoting oral health should be encouraged as an indispensable part of a healthy lifestyle to reduce the burden of global chronic noncommunicable and communicable diseases[80], which was an initiative of the World Health Assembly as early as 2007. There is currently no doubt that periodontitis is certainly preventable and controllable, as is P. gingivalis, and should be regarded as a modifiable factor for diseases. Reducing the load of P. gingivalis through periodontal intervention has potential benefits for oral health and a direct or indirect positive impact on overall health, even if it prevents the possibility of such a connection. We should be aware of the risk and address it as soon as possible to avoid threatening our health. Moreover, interrelations between P. gingivalis and other oral pathogens (such as Actinobacillus actinomycetemcomitans and Fusobacterium nucleatum), together with gut microbiota, are another subject of intensive study.
References
- Özlem Yilmaz; The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology 2008, 154, 2897-2903, 10.1099/mic.0.2008/021220-0.
- Ingar Olsen; John D. Lambris; George Hajishengallis; Porphyromonas gingivalis disturbs host–commensal homeostasis by changing complement function. Journal of Oral Microbiology 2017, 9, 1340085-1340085, 10.1080/20002297.2017.1340085.
- Aditi Chopra; Subraya G. Bhat; Karthik Sivaraman; Porphyromonas gingivalis adopts intricate and unique molecular mechanisms to survive and persist within the host: a critical update. Journal of Oral Microbiology 2020, 12, 1801090, 10.1080/20002297.2020.1801090.
- Morten Enersen; Kazuhiko Nakano; Atsuo Amano; Porphyromonas gingivalis fimbriae. Journal of Oral Microbiology 2013, 5, -, 10.3402/jom.v5i0.20265.
- Weizhe Xu; Wei Zhou; Huizhi Wang; Shuang Liang; Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Therapeutic Proteins and Peptides 2020, 120, 45-84, 10.1016/bs.apcsb.2019.12.001.
- E. Jeong; J-Y. Lee; S-J. Kim; Jeomil Choi; Predominant immunoreactivity ofPorphyromonas gingivalisheat shock protein in autoimmune diseases. Journal of Periodontal Research 2012, 47, 811-816, 10.1111/j.1600-0765.2012.01501.x.
- Yonghua Guo; Ky-Anh Nguyen; Jan Potempa; Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2000 2010, 54, 15-44, 10.1111/j.1600-0757.2010.00377.x.
- M.J. Gui; Stuart G Dashper; Nada Slakeski; Y.-Y. Chen; Eric C. Reynolds; Spheres of influence:Porphyromonas gingivalisouter membrane vesicles. Molecular Oral Microbiology 2015, 31, 365-378, 10.1111/omi.12134.
- Hua Xie; Biogenesis and function ofPorphyromonas gingivalisouter membrane vesicles. Future Microbiology 2015, 10, 1517-1527, 10.2217/fmb.15.63.
- Lone Forner; Tove Larsen; Mogens Kilian; Palle Holmstrup; Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. Journal of Clinical Periodontology 2006, 33, 401-407, 10.1111/j.1600-051x.2006.00924.x.
- Ellen Berggreen; H. Wiig; Lymphangiogenesis and Lymphatic Function in Periodontal Disease. Journal of Dental Research 2013, 92, 1074-1080, 10.1177/0022034513504589.
- Emil V. Kozarov; Brian R. Dorn; Charles E. Shelburne; William A. Dunn; Ann Progulske-Fox; Human Atherosclerotic Plaque Contains Viable InvasiveActinobacillus actinomycetemcomitansandPorphyromonas gingivalis. Arteriosclerosis, Thrombosis, and Vascular Biology 2005, 25, e17-e18, 10.1161/01.atv.0000155018.67835.1a.
- Małgorzata Benedyk; Piotr Mateusz Mydel; Nicolas Delaleu; Karolina Płaza; Katarzyna Gawron; Aleksandra Milewska; Katarzyna Maresz; Joanna Koziel; Krzysztof Pyrc; Jan Potempa; et al. Gingipains: Critical Factors in the Development of Aspiration Pneumonia Caused by Porphyromonas gingivalis. Journal of Innate Immunity 2015, 8, 185-198, 10.1159/000441724.
- Kumiko Yamatake; Maki Maeda; Tomoko Kadowaki; Ryosuke Takii; Takayuki Tsukuba; Takashi Ueno; Eiki Kominami; Sadaki Yokota; Kenji Yamamoto; Role for Gingipains in Porphyromonas gingivalis Traffic to Phagolysosomes and Survival in Human Aortic Endothelial Cells. Infection and Immunity 2007, 75, 2090-2100, 10.1128/iai.01013-06.
- JebaMercy Gnanasekaran; Adi Binder Gallimidi; Elias Saba; Karthikeyan Pandi; Luba Eli Berchoer; Esther Hermano; Sarah Angabo; Hasna′a Makkawi; Arin Khashan; Alaa Daoud; et al.Michael ElkinGabriel Nussbaum Intracellular Porphyromonas gingivalis Promotes the Tumorigenic Behavior of Pancreatic Carcinoma Cells. Cancers 2020, 12, 2331, 10.3390/cancers12082331.
- Ahmed R. El-Awady; Brodie Miles; Elizabeth Scisci; Zoya B. Kurago; Chithra D. Palani; Roger M. Arce; Jennifer L. Waller; Caroline A. Genco; Connie Slocum; Matthew Manning; et al.Patricia V. SchoenleinChristopher W Cutler Porphyromonas gingivalis Evasion of Autophagy and Intracellular Killing by Human Myeloid Dendritic Cells Involves DC-SIGN-TLR2 Crosstalk. PLOS Pathogens 2015, 11, e1004647, 10.1371/journal.ppat.1004647.
- Denis F. Kinane; Panagiota G. Stathopoulou; Panos N. Papapanou; Periodontal diseases. Nature Reviews Disease Primers 2017, 3, nrdp201738, 10.1038/nrdp.2017.38.
- Li Li; Emmanuel Messas; Eraldo L Batista Jr; Robert A. Levine; Salomon Amar; Porphyromonas gingivalisInfection Accelerates the Progression of Atherosclerosis in a Heterozygous Apolipoprotein E–Deficient Murine Model. Circulation 2002, 105, 861-867, 10.1161/hc0702.104178.
- Jia-Shun Wu; Min Zheng; Mei Zhang; Xin Pang; Li Li; Sha-Sha Wang; Xiao Yang; Jing-Biao Wu; Ya-Ling Tang; Xin-Hua Liang; et al. Porphyromonas gingivalis Promotes 4-Nitroquinoline-1-Oxide-Induced Oral Carcinogenesis With an Alteration of Fatty Acid Metabolism. Frontiers in Microbiology 2018, 9, 2081, 10.3389/fmicb.2018.02081.
- Stephen S. Dominy; Casey Lynch; Florian Ermini; Malgorzata Benedyk; Agata Marczyk; Andrei Konradi; Mai Nguyen; Ursula Haditsch; Debasish Raha; Christina Griffin; et al.Leslie J. HolsingerShirin Arastu-KapurSamer KabaAlexander LeeMark I. RyderBarbara PotempaPiotr MydelAnnelie HellvardKarina AdamowiczHatice HasturkGlenn D. WalkerEric C. ReynoldsRichard L. M. FaullMaurice A. CurtisMike DragunowJan Potempa Porphyromonas gingivalisin Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Science Advances 2019, 5, eaau3333, 10.1126/sciadv.aau3333.
- Mengru Xie; Qingming Tang; Jiaming Nie; Chao Zhang; Xin Zhou; Shaoling Yu; Jiwei Sun; Xiang Cheng; Nianguo Dong; Yu Hu; et al.Lili Chen BMAL1-Downregulation Aggravates Porphyromonas Gingivalis -Induced Atherosclerosis by Encouraging Oxidative Stress. Circulation Research 2020, 126, e15-e29, 10.1161/circresaha.119.315502.
- Myriam Bélanger; Paulo H. Rodrigues; Jr. William A. Dunn; Ann Progulske-Fox; Autophagy: A Highway for Porphyromonas gingivalis in Endothelial Cells. Autophagy 2006, 2, 165-170, 10.4161/auto.2828.
- Bin Liu; Lan Cheng; Dali Liu; Jia Wang; Xiuli Zhang; Rong Shu; Jingping Liang; Role of p38 Mitogen-Activated Protein Kinase Pathway inPorphyromonas gingivalisLipopolysaccharide–Induced VCAM-1 Expression in Human Aortic Endothelial Cells. Journal of Periodontology 2012, 83, 955-962, 10.1902/jop.2011.110406.
- Mary Khlgatian; Hamdy Nassar; Hsin-Hua Chou; Frank C. Gibson; Caroline Attardo Genco; Fimbria-dependent activation of cell adhesion molecule expression in Porphyromonas gingivalis-infected endothelial cells.. Infection and Immunity 2002, 70, -.
- Georg A Roth; Bernhard Moser; S. J. Huang; Justin S. Brandt; Y. Huang; Panos N. Papapanou; Ann Marie Schmidt; E. Lalla; Infection with a periodontal pathogen induces procoagulant effects in human aortic endothelial cells. Journal of Thrombosis and Haemostasis 2006, 4, 2256-2261, 10.1111/j.1538-7836.2006.02128.x.
- Koji Nakayama; Porphyromonas gingivalis cell-induced hemagglutination and platelet aggregation. Periodontology 2000 2010, 54, 45-52, 10.1111/j.1600-0757.2010.00351.x.
- Harvey A. Schenkein; Panos N. Papapanou; Robert Genco; Mariano Sanz; Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontology 2000 2020, 83, 90-106, 10.1111/prd.12304.
- A. Pollreisz; Y. Huang; Georg A Roth; B. Cheng; Moritz Kebschull; P. N. Papapanou; A. M. Schmidt; E. Lalla; Enhanced monocyte migration and pro-inflammatory cytokine production byPorphyromonas gingivalisinfection. Journal of Periodontal Research 2010, 45, 239-245, 10.1111/j.1600-0765.2009.01225.x.
- Xiu-Ying Li; Chun Wang; Xue-Rong Xiang; Fang-Chun Chen; Cong-Ming Yang; Jing Wu; Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncology Reports 2013, 30, 1329-1336, 10.3892/or.2013.2600.
- D.J. Kim; J.H. Rho; B.H. Woo; Ji-Young Joo; J.M. Song; Hae Ryoun Park; J.Y. Lee; J.H. Lee; Periodontal Pathogens Modulate Lipid Flux via Fatty Acid Binding Protein 4. Journal of Dental Research 2019, 98, 1511-1520, 10.1177/0022034519880824.
- Paul M. Brown; David J. Kennedy; Richard E. Morton; Maria Febbraio; CD36/SR-B2-TLR2 Dependent Pathways Enhance Porphyromonas gingivalis Mediated Atherosclerosis in the Ldlr KO Mouse Model. PLOS ONE 2015, 10, e0125126-e0125126, 10.1371/journal.pone.0125126.
- Johanna Lönn; S. Ljunggren; K. Klarström-Engström; I. Demirel; T. Bengtsson; H. Karlsson; Lipoprotein modifications by gingipains ofPorphyromonas gingivalis. Journal of Periodontal Research 2018, 53, 403-413, 10.1111/jre.12527.
- Guirong Liu; Jing Deng; Qiang Zhang; Wenbin Song; Shulan Chen; Xiuxiu Lou; Pengmei Zhang; Keqing Pan; Porphyromonas gingivalisLipopolysaccharide Stimulation of Vascular Smooth Muscle Cells Activates Proliferation and Calcification. Journal of Periodontology 2016, 87, 828-836, 10.1902/jop.2016.150602.
- Wen Wei Yang; Bin Guo; Wen Yuan Jia; Yafei Wu; Porphyromonas gingivalis-derived outer membrane vesicles promote calcification of vascular smooth muscle cells through ERK1/2-RUNX2. FEBS Open Bio 2016, 6, 1310-1319, 10.1002/2211-5463.12151.
- Koichiro Wada; Yoshinori Kamisaki; Molecular dissection of Porphyromonas gingivalis-related arteriosclerosis: a novel mechanism of vascular disease. Periodontology 2000 2010, 54, 222-234, 10.1111/j.1600-0757.2009.00336.x.
- Lili Ye; Yinhua Jiang; Weidong Liu; Haibiao Tao; Correlation between periodontal disease and oral cancer risk: A meta-analysis. Journal of Cancer Research and Therapeutics 2016, 12, 237-C240, 10.4103/0973-1482.200746.
- L Wen; W Mu; H Lu; X Wang; J Fang; Y Jia; Q Li; D Wang; S Wen; J Guo; et al.W DaiX RenJ CuiG ZengJ GaoZ WangB Cheng Porphyromonas gingivalis Promotes Oral Squamous Cell Carcinoma Progression in an Immune Microenvironment.. null 2020, 22034520909312, -.
- Jeffrey N Katz; Mairelys D Onate; Kaleb M Pauley; Indraneel Bhattacharyya; Seunghee Cha; Presence of Porphyromonas gingivalis in gingival squamous cell carcinoma. International Journal of Oral Science 2011, 3, 209-215, 10.4248/ijos11075.
- Jungnam Lee; JoAnn S. Roberts; Kalina R. Atanasova; Nityananda Chowdhury; Kyudong Han; Özlem Yilmaz; Human Primary Epithelial Cells Acquire an Epithelial-Mesenchymal-Transition Phenotype during Long-Term Infection by the Oral Opportunistic Pathogen, Porphyromonas gingivalis. Frontiers in Cellular and Infection Microbiology 2017, 7, 493-493, 10.3389/fcimb.2017.00493.
- Jun Ohshima; Qian Wang; Zackary R. Fitzsimonds; Daniel P. Miller; Maryta N. Sztukowska; Young-Jung Jung; Mikako Hayashi; Marvin Whiteley; Richard J. Lamont; Streptococcus gordoniiprograms epithelial cells to resist ZEB2 induction byPorphyromonas gingivalis. Proceedings of the National Academy of Sciences 2019, 116, 8544-8553, 10.1073/pnas.1900101116.
- Ingar Olsen; Özlem Yilmaz; Possible role of Porphyromonas gingivalis in orodigestive cancers. Journal of Oral Microbiology 2019, 11, 1563410, 10.1080/20002297.2018.1563410.
- Song Mao; Yoonsuk Park; Yoshiaki Hasegawa; Gena D. Tribble; Chlöe E. James; Martin Handfield; M. Franci Stavropoulos; Özlem Yilmaz; Richard J. Lamont; Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cellular Microbiology 2007, 9, 1997-2007, 10.1111/j.1462-5822.2007.00931.x.
- L. Yao; C. Jermanus; B. Barbetta; C. Choi; P. Verbeke; D.M. Ojcius; Ö. Yilmaz; Porphyromonas gingivalisinfection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Molecular Oral Microbiology 2010, 25, 89-101, 10.1111/j.2041-1014.2010.00569.x.
- Catherine E. Moffatt; Richard J. Lamont; Porphyromonas gingivalis Induction of MicroRNA-203 Expression Controls Suppressor of Cytokine Signaling 3 in Gingival Epithelial Cells. Infection and Immunity 2011, 79, 2632-2637, 10.1128/iai.00082-11.
- Jungnam Lee; JoAnn S. Roberts; Kalina R. Atanasova; Nityananda Chowdhury; Ö. Yilmaz; A novel kinase function of a nucleoside-diphosphate-kinase homologue in Porphyromonas gingivalis is critical in subversion of host cell apoptosis by targeting heat-shock protein 27. Cellular Microbiology 2018, 20, e12825, 10.1111/cmi.12825.
- Ö. Yilmaz; Luyu Yao; Kazuhiko Maeda; Timothy M. Rose; Emma L. Lewis; Memed Duman; Richard J. Lamont; David M. Ojcius; ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cellular Microbiology 2007, 10, 863-875, 10.1111/j.1462-5822.2007.01089.x.
- Masae Kuboniwa; Yoshiaki Hasegawa; Song Mao; Satoshi Shizukuishi; Atsuo Amano; Richard J. Lamont; Özlem Yilmaz; P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes and Infection 2008, 10, 122-128, 10.1016/j.micinf.2007.10.011.
- Qian Wang; Maryta Sztukowska; Akintunde Ojo; David A. Scott; Huizhi Wang; Richard J. Lamont; FOXO responses toPorphyromonas gingivalisin epithelial cells. Cellular Microbiology 2015, 17, 1605-1617, 10.1111/cmi.12459.
- Sabine Groeger; F. Denter; G. Lochnit; M. L. Schmitz; J. Meyle; Porphyromonas gingivalis Cell Wall Components Induce Programmed Death Ligand 1 (PD-L1) Expression on Human Oral Carcinoma Cells by a Receptor-Interacting Protein Kinase 2 (RIP2)-Dependent Mechanism. Infection and Immunity 2020, 88, -, 10.1128/iai.00051-20.
- Ana Carolina Morandini; Erivan Schnaider Ramos-Junior; Jan Potempa; Ky-Anh Nguyen; Ana Carolina Oliveira; Maria Bellio; David M. Ojcius; Julio Scharfstein; Robson Coutinho-Silva; Porphyromonas gingivalis Fimbriae Dampen P2X7-Dependent Interleukin-1β Secretion. Journal of Innate Immunity 2014, 6, 831-845, 10.1159/000363338.
- Chunrong Chang; Hongyan Wang; Junchao Liu; Chunling Pan; Ngmei Zhang; Xin Li; Yaping Pan; Porphyromonas gingivalis Infection Promoted the Proliferation of Oral Squamous Cell Carcinoma Cells through the miR-21/PDCD4/AP-1 Negative Signaling Pathway.. ACS Infectious Diseases 2019, 5, 1336-1347, 10.1021/acsinfecdis.9b00032.
- Na Hee Ha; Bok Hee Woo; Da Jeong Kim; Eun Sin Ha; Jeomil Choi; Sung Jo Kim; Bong Soo Park; Ji Hye Lee; Hae Ryoun Park; Prolonged and repetitive exposure to Porphyromonas gingivalis increases aggressiveness of oral cancer cells by promoting acquisition of cancer stem cell properties. Tumor Biology 2015, 36, 9947-9960, 10.1007/s13277-015-3764-9.
- Hiroaki Inaba; Hideyuki Sugita; Masae Kuboniwa; Soichi Iwai; Masakazu Hamada; Takeshi Noda; Ichijiro Morisaki; Richard J. Lamont; Atsuo Amano; Porphyromonas gingivalispromotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cellular Microbiology 2013, 16, 131-145, 10.1111/cmi.12211.
- Na Hee Ha; Dae Gun Park; Bok Hee Woo; Da Jeong Kim; Jeomil Choi; Bong Soo Park; Yong-Deok Kim; Jihye Lee; Hae Ryoun Park; Porphyromonas gingivalis increases the invasiveness of oral cancer cells by upregulating IL-8 and MMPs. Cytokine 2016, 86, 64-72, 10.1016/j.cyto.2016.07.013.
- Wei-Long Zhang; Sha-Sha Wang; Hao-Fan Wang; Ya-Jie Tang; Yaling Tang; Xin-Hua Liang; Who is who in oral cancer?. Experimental Cell Research 2019, 384, 111634, 10.1016/j.yexcr.2019.111634.
- Chang-Kai Chen; Yung-Tsan Wu; Y.-C. Chang; Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched-cohort study. Alzheimer's Research & Therapy 2017, 9, 1-7, 10.1186/s13195-017-0282-6.
- Sophie Poole; Sim K. Singhrao; Lakshmyya Kesavalu; Michael A. Curtis; StJohn Crean; Determining the Presence of Periodontopathic Virulence Factors in Short-Term Postmortem Alzheimer's Disease Brain Tissue. Journal of Alzheimer's Disease 2013, 36, 665-677, 10.3233/jad-121918.
- Mariagrazia Pizza; Dominy Ss; Lynch C; Ermini F; Benedyk M; Marczyk A; Konradi A; Nguyen M; Haditsch U; Raha D; et al.Griffin CHolsinger LjArastu-Kapur SKaba SLee ARyder MiPotempa BMydel PHellvard AAdamowicz KHasturk HWalker GdReynolds EcFaull RlmCurtis MaDragunow MPotempa J Faculty Opinions recommendation of Porphyromonas gingivalis in Alzheimer's disease brains: Evidence for disease causation and treatment with small-molecule inhibitors.. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2019, 5, eaau3333, 10.3410/f.734906096.793558207.
- Zhou Wu; Junjun Ni; Yicong Liu; Jessica L. Teeling; Fumiko Takayama; Alex Collcutt; Paul Ibbett; Hiroshi Nakanishi; Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain, Behavior, and Immunity 2017, 65, 350-361, 10.1016/j.bbi.2017.06.002.
- Jing Zhang; Chunbo Yu; Xuan Zhang; Huiwen Chen; Jiachen Dong; Weili Lu; Zhongchen Song; Wei Zhou; Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. Journal of Neuroinflammation 2018, 15, 1-14, 10.1186/s12974-017-1052-x.
- Ran Nie; Zhou Wu; Junjun Ni; Fan Zeng; Weixian Yu; Yufeng Zhang; Tomoko Kadowaki; Haruhiko Kashiwazaki; Jessica L. Teeling; Yanmin Zhou; et al. Porphyromonas gingivalis Infection Induces Amyloid-β Accumulation in Monocytes/Macrophages. Journal of Alzheimer's Disease 2019, 72, 479-494, 10.3233/jad-190298.
- Ingar Olsen; Sim K. Singhrao; Interaction between genetic factors, Porphyromonas gingivalis and microglia to promote Alzheimer’s disease. Journal of Oral Microbiology 2020, 12, 1820834, 10.1080/20002297.2020.1820834.
- Atsuo Amano; Ichiro Nakagawa; Nobuo Okahashi; Nobushiro Hamada; Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. Journal of Periodontal Research 2004, 39, 136-142, 10.1111/j.1600-0765.2004.00719.x.
- Mazen M. Jamil Al-Obaidi; M.N.M. Desa; Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial–Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cellular and Molecular Neurobiology 2018, 38, 1349-1368, 10.1007/s10571-018-0609-2.
- Yicong Liu; Zhou Wu; Yurika Nakanishi; Junjun Ni; Yoshinori Hayashi; Fumiko Takayama; Yanmin Zhou; Tomoko Kadowaki; Hiroshi Nakanishi; Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Scientific Reports 2017, 7, 1-13, 10.1038/s41598-017-12173-1.
- Ingar Olsen; Martin A. Taubman; Sim K. Singhrao; Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. Journal of Oral Microbiology 2016, 8, 33029, 10.3402/jom.v8.33029.
- Filippo Graziani; Stefano Gennai; Anna Solini; Morena Petrini; A systematic review and meta-analysis of epidemiologic observational evidence on the effect of periodontitis on diabetes An update of the EFP-AAP review. Journal of Clinical Periodontology 2017, 45, 167-187, 10.1111/jcpe.12837.
- Vincent Blasco-Baque; Lucile Garidou; Céline Pomié; Quentin Escoula; Pascale Loubieres; Sandrine Le Gall-David; Mathieu Lemaitre; Simon Nicolas; Pascale Klopp; Aurélie Waget; et al.Vincent AzalbertAndré ColomMartine Bonnaure-MalletPhilippe KemounMatteo SerinoRémy Burcelin Periodontitis induced byPorphyromonas gingivalisdrives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut 2016, 66, 872-885, 10.1136/gutjnl-2015-309897.
- Naoki Sasaki; Sayaka Katagiri; Rina Komazaki; Kazuki Watanabe; Shogo Maekawa; Takahiko Shiba; Sayuri Udagawa; Yasuo Takeuchi; Anri Ohtsu; Takashi Kohda; et al.Haruka ToharaNaoyuki MiyasakaTomomitsu HirotaMayumi TamariYuichi Izumi Endotoxemia by Porphyromonas gingivalis Injection Aggravates Non-alcoholic Fatty Liver Disease, Disrupts Glucose/Lipid Metabolism, and Alters Gut Microbiota in Mice. Frontiers in Microbiology 2018, 9, 2470, 10.3389/fmicb.2018.02470.
- Mariko Seyama; Kaya Yoshida; Natsumi Fujiwara; Kisho Ono; Takanori Eguchi; Hotaka Kawai; Jiajie Guo; Yao Weng; Yuan Haoze; Kenta Uchibe; et al.Mika IkegameAkira SasakiHitoshi NagatsukaKuniaki OkamotoHirohiko OkamuraKazumi Ozaki Outer membrane vesicles of Porphyromonas gingivalis attenuate insulin sensitivity by delivering gingipains to the liver. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2020, 1866, 165731, 10.1016/j.bbadis.2020.165731.
- J. Tian; C. Liu; X. Zheng; X. Jia; X. Peng; R. Yang; X. Zhou; Xin Xu; Porphyromonas gingivalis Induces Insulin Resistance by Increasing BCAA Levels in Mice. Journal of Dental Research 2020, 99, 839-846, 10.1177/0022034520911037.
- Kei Arimatsu; Hiroshi Yamada; Haruna Miyazawa; Takayoshi Minagawa; Masahiro Nakajima; Mark I. Ryder; Kazuyoshi Gotoh; Daisuke Motooka; Shigeki Nakamura; Tadayuki Iida; et al.Kazuhisa Yamazaki Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Scientific Reports 2014, 4, 4828, 10.1038/srep04828.
- Aruna D. Pradhan; JoAnn E. Manson; Nader Rifai; Julie E. Buring; Paul M. Ridker; C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus. JAMA 2001, 286, 327-334, 10.1001/jama.286.3.327.
- Fanny Le Sage; Olivier Meilhac; Marie-Paule Gonthier; Porphyromonas gingivalis lipopolysaccharide induces pro-inflammatory adipokine secretion and oxidative stress by regulating Toll-like receptor-mediated signaling pathways and redox enzymes in adipocytes. Molecular and Cellular Endocrinology 2017, 446, 102-110, 10.1016/j.mce.2017.02.022.
- Ryan T Demmer; Jr D.R. Jacobs; Ranjana Singh; Aleksandra M Zuk; Marlon S Rosenbaum; Panos N Papapanou; Moise Desvarieux; Periodontal Bacteria and Prediabetes Prevalence in ORIGINS. Journal of Dental Research 2015, 94, 201S-211S, 10.1177/0022034515590369.
- Uppoor G. Bhat; Vladimir Ilievski; Terry G. Unterman; Keiko Watanabe; Porphyromonas gingivalisLipopolysaccharide Upregulates Insulin Secretion From Pancreatic β Cell Line MIN6. Journal of Periodontology 2014, 85, 1629-1636, 10.1902/jop.2014.140070.
- V. Ilievski; U. G. Bhat; S. Suleiman-Ata; B. A. Bauer; P. T. Toth; S. T. Olson; T. G. Unterman; K. Watanabe; Oral application of a periodontal pathogen impacts SerpinE1 expression and pancreatic islet architecture in prediabetes. Journal of Periodontal Research 2017, 52, e12474-1041, 10.1111/jre.12474.
- Haruna Takamura; Kaya Yoshida; Hirohiko Okamura; Natsumi Fujiwara; Kazumi Ozaki; Porphyromonas gingivalis attenuates the insulin-induced phosphorylation and translocation of forkhead box protein O1 in human hepatocytes. Archives of Oral Biology 2016, 69, 19-24, 10.1016/j.archoralbio.2016.05.010.
- Hiroki Takeuchi; Naoko Sasaki; Shunsuke Yamaga; Masae Kuboniwa; Michiya Matsusaki; Atsuo Amano; Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of junctional adhesion molecule 1.. PLOS Pathogens 2019, 15, e1008124, 10.1371/journal.ppat.1008124.
- Poul Erik Petersen; World Health Organization global policy for improvement of oral health - World Health Assembly 2007. International Dental Journal 2008, 58, 115-121, 10.1111/j.1875-595x.2008.tb00185.x.

