Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Margherita Sisto | -- | 2375 | 2022-10-31 17:53:55 | | | |
| 2 | Rita Xu | Meta information modification | 2375 | 2022-11-01 03:34:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sisto, M.; Ribatti, D.; Lisi, S. Sjögren’s Syndrome Pathogenic Molecular Pathways. Encyclopedia. Available online: https://encyclopedia.pub/entry/32133 (accessed on 26 July 2026).
Sisto M, Ribatti D, Lisi S. Sjögren’s Syndrome Pathogenic Molecular Pathways. Encyclopedia. Available at: https://encyclopedia.pub/entry/32133. Accessed July 26, 2026.
Sisto, Margherita, Domenico Ribatti, Sabrina Lisi. "Sjögren’s Syndrome Pathogenic Molecular Pathways" Encyclopedia, https://encyclopedia.pub/entry/32133 (accessed July 26, 2026).
Sisto, M., Ribatti, D., & Lisi, S. (2022, October 31). Sjögren’s Syndrome Pathogenic Molecular Pathways. In Encyclopedia. https://encyclopedia.pub/entry/32133
Sisto, Margherita, et al. "Sjögren’s Syndrome Pathogenic Molecular Pathways." Encyclopedia. Web. 31 October, 2022.
Copy Citation
Sjögren’s syndrome (SS) is a systemic autoimmune rheumatic disorder characterized by the lymphocytic infiltration of exocrine glands and the production of autoantibodies to self-antigens. The involvement of the exocrine glands drives the pathognomonic manifestations of dry eyes (keratoconjunctivitis sicca) and dry mouth (xerostomia) that define sicca syndrome.
Sjögren’s syndrome
autoimmunity
apoptosis
aquaporin
1. Introduction
Sjögren’s syndrome (SS) is a multifactorial systemic autoimmune disease, the pathophysiology of which has not yet been fully deciphered [1][2] (Figure 1). SS is characterized by a wide spectrum of clinical manifestations and marked exocrine gland dysfunction. SS is classified as primary SS (pSS) when the clinical manifestations occur alone or as secondary SS when associated with another autoimmune disease [1][2].
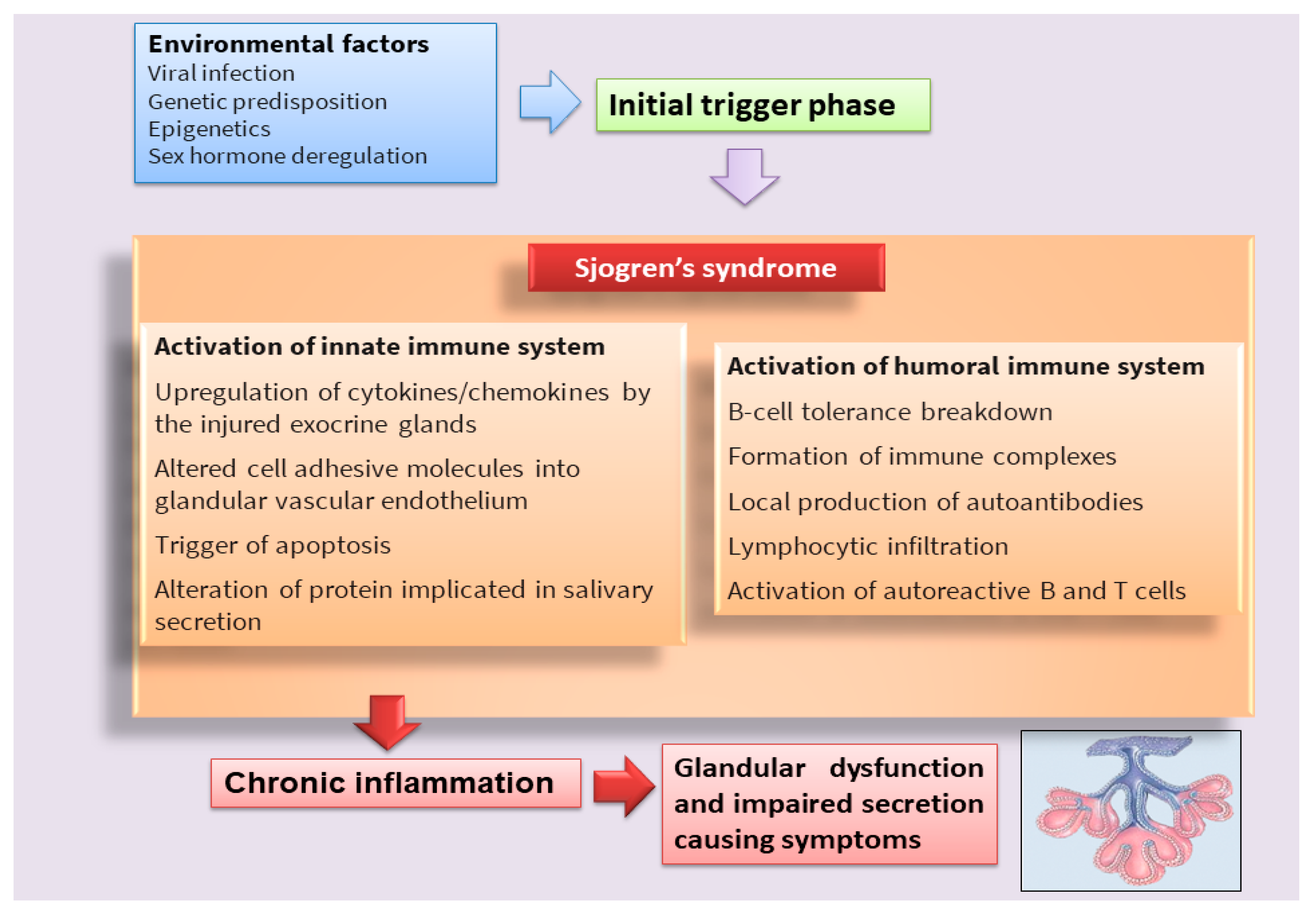
Figure 1. Scheme that clarifies the hypothetical onset of SS.
Classically, it has been postulated that sicca symptoms in SS patients are a two-step process whereby the lymphocytic infiltration of the lacrimal and salivary glands (SG) is followed by epithelial cell destruction, resulting in keratoconjunctivitis sicca and xerostomia [3]. Recently, great efforts have been made to elucidate the mechanisms involved in the pathogenesis of the disease in order to identify potential new therapeutic targets in SS (Table 1). In recent years, interesting discoveries have shown that pSS has pathogenic mechanisms and etiology in common with other autoimmune diseases that predominantly afflict women, represented by rheumatoid arthritis (RA) and systemic lupus erythematosus, which preferentially affect specific target organs. Indeed, these autoimmune diseases, characterized by a chronic inflammatory condition, show similar clinical manifestations, serological profiles, and immunological alterations. Currently, the term poly-autoimmunity is used to indicate the co-existence of these three pathologies in the same patient, and, sometimes, these conditions can also be manifested by members belonging to the same family. This suggests that the molecular mechanisms underlying the onset of these pathologies could be the same, and elucidating these mechanisms in pSS could be of help in guiding researchers toward understanding the pathogenesis of related autoimmune diseases [4].
Table 1. Schematic illustration of therapies used in SS.
| Nomenclatures of Therapies | Definition | Examples |
|---|---|---|
| Thopical therapies | Interventions directly applied to the mucosal surface involved | Saliva substitutes, ocular tears, ocular gels/ointment |
| Systemic therapies | Drugs administered orally or intravenously for systemic disease | Antimalarials, glucocorticoids, immunosuppressive agents, intravenous immunoglobulins, biologics drugs |
| Systemic therapies for severe refractory diseases | Drugs administered intravenously | B-cells targeted therapies |
2. Recent Advances in Apoptosis in SS
Over the last several years, an increasing number of studies have revealed the key role of apoptosis, or programmed cell death (PCD), in the pathogenesis of SS [5][6]. Apoptosis is a critical process, highly complex and sophisticated, that is conserved throughout evolution, development, and aging to ensure both physiological and morphological changes as well as the elimination of damaged cells [7]. The mechanism of apoptosis is implicated in a cascade of molecular events that lead to the development of autoimmune disorders [8]. In the last few decades, apoptosis has been hypothesized as a mechanism of cell death in the SGs of pSS patients on the basis of data collected using experimental SS mouse models [9], pointing to glandular epithelial cells as active players in this mechanism [10]. In fact, pSS patients present increased apoptosis in the salivary glandular epithelium and show the co-localization of Fas (Apo-1/CD95) antigen and Fas ligand (FasL) in ductal and acinar cells, suggesting that the trigger of the apoptotic cascade occurs through the Fas/FasL system [11]. On the other hand, cytotoxic T cells have also been reported to play a crucial role in apoptotic events in the SS salivary epithelium. Indeed, cytotoxic T cells surround the epithelial cells following inflammatory responses and, through Fas ligand interactions and perforin and granzyme release, promote apoptosis in the pSS epithelium, thus leading to advanced glandular tissue destruction and chronic sialadenitis [12][13].
It is reported that there are multiple pathways involved in the apoptotic mechanism in SS. One of the best-known pathways that determine cell fate and evolution toward apoptosis is mediated by caspase cascade activation via the extrinsic or intrinsic mechanism (Figure 2). Caspases cleave a variety of protein substrates within the cell, including alpha-fodrin [14][15] and Ro and La proteins [5][16], and, therefore, it is conceivable that epithelial cell apoptosis may provide cellular proteins as autoantigens that then perpetuate the autoimmune response in SS [17][18][19].
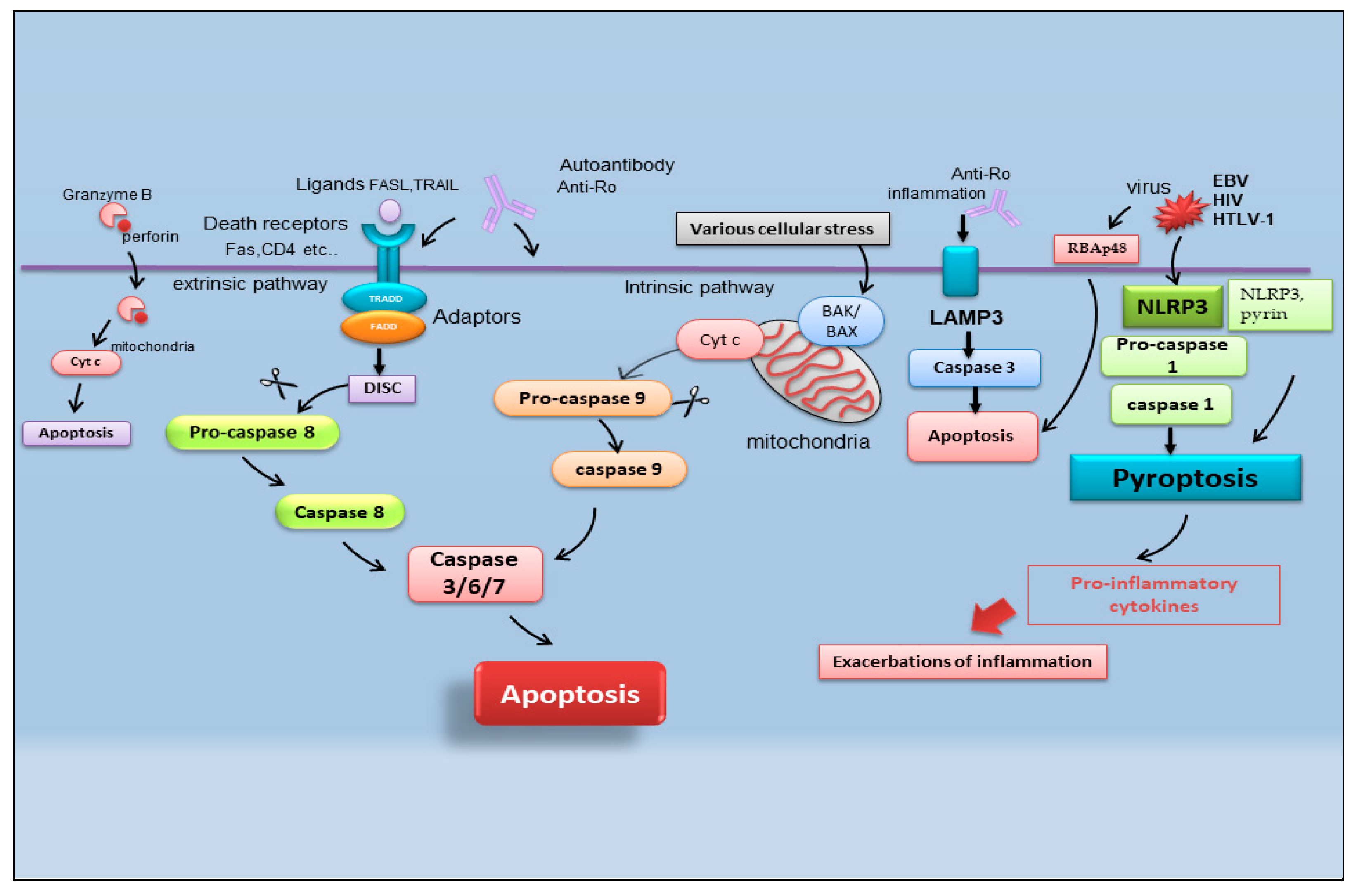
Figure 2. Schematic representation of recently identified apoptotic events in SS. B-cell lymphoma protein 2 antagonist killer 1 (BAK); B-cell lymphoma protein-2-associated X protein (BAX); cytochrome c (Cyt c); death-inducing signaling complex (DISC); Epstein–Barr virus (EBV); fatty acid synthetase ligand (FASL); Fas-associated protein with death domain (FADD); human immunodeficiency virus (HIV); human T-cell leukemia virus type 1 (HTLV-1); lysosome-associated membrane protein 3 (LAMP3); nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3); tumor necrosis factor receptor type 1-associated DEATH domain (TRADD); TNF-related apoptosis-inducing ligand (TRAIL) retinoblastoma-associated protein 48 (RBAp48).
2.1. Pyroptosis
In the field of SS apoptosis research, recently, the excessive exacerbation of pyroptosis has been proven to play a crucial role. Pyroptosis is an overactive PCD characterized by pore formation in cell membranes, cell rupture, and the release of intracellular contents and pro-inflammatory cytokines, such as IL-1β and IL-18. This prolonged release of inflammatory factors drives the overactivation of the immune system, promoting autoimmunity [20]. The discovery of the activation of pyroptosis in SS creates a connecting bridge with the activation of cytotoxic T cells observed in SS, as, under this condition of the uncontrolled release of pro-inflammatory cytokines, autoantibodies and/or autoreactive T cells can uncontrollably attack the body, causing autoimmune diseases [21]. Among the canonical inflammasomes that enable the induction of pyroptosis (NLRP1, NLRP3, NLRC4, interferon-inducible protein AIM2, and pyrin) [22], the increased expression of NLRP3 (NOD (nucleotide oligomerization domain)-, LRR (leucine-rich repeat)-, and PYD (pyrin domain)-containing protein 3) inflammasome-related elements in peripheral blood mononuclear cells or macrophages infiltrating the SGs of pSS patients was detected, determined by inflammatory circulating cell-free DNA accumulated in the SS patients’ serum [23]. Similarly, the accumulation of damaged cytoplasmic DNA in the SG ductal cells of patients with pSS was demonstrated to activate the AIM2 inflammasome, causing the intensive expression of pyroptosomes in the SG tissue [24]. Pyroptosis is closely associated with the activation of caspase-dependent cascades, and, in SS, type I IFN upregulated the expression of caspase-1 in pSS epithelial cells (SGECs) and may accelerate NLRP3 or AMI2 inflammasome-associated pyroptosis [25].
2.2. Apoptosis and Viral Infection in SS
Interestingly, pyroptosis, driving CD4 T-cell depletion in HIV-1 infection [26], could be one of the molecular mechanisms involving viral proteins in the etiopathogenesis of pSS [27]. The role of viral infections in the pathogenesis of SS represents an important line of research carried out by the Nakamura group, demonstrating that viruses could essentially change the expression or regulation of various genes, including those that regulate apoptosis [28]. In pSS, the initial apoptosis of epithelial cells may be a normal response to viral infection, but the inability to regulate the apoptotic process may then perpetuate epithelial cell impairment and the resultant cellular and humoral features that characterize SS. In fact, viral infections, such as Epstein–Barr virus (EBV) and human T-cell leukemia virus type 1 (HTLV-1), change the phenotype or features of SS SGECs through the breakdown of local immunological tolerance and determine the activation of the apoptotic cascade [28].
Indeed, there are recent insights regarding the relationship between apoptosis and viral infection in SS. Increased retinoblastoma-associated protein 48 (RBAp48) expression, which controls chromatin organization induced by viral infections such as HIV (human immunodeficiency virus), was also found in the epithelial cells of minor SGs of labial biopsies from patients affected by pSS, in which the cells undergo the apoptotic process [29]. Therefore, the RBAp48 of SGs cells in mice and humans was reported to be upregulated by estrogen deficiency, which triggered apoptosis in the target cells [29][30][31]. Although the relationship between viral infection and autoantigen formation in SS is shrouded in mystery, apoptosis in target cells due to viral infection can induce the upregulation of multiple enzymatic activities to generate pathogenic epitopes from intracellular molecules, leading to an autoimmune response [31].
2.3. Lysosome-Associated Membrane Protein 3-Dependent Apoptosis
Another crucial point in the pathogenesis of SS is the upregulation of lysosome-associated membrane protein 3 (LAMP3), a membrane glycoprotein predominantly localized in lysosomes induced by IFN. An interesting recent study [32] demonstrated the increased expression of LAMP3 in a subset of pSS cases. The stratification of patients based on their clinical features suggested a link between increased LAMP3 expression and the presence of serum autoantibodies, including anti-Ro/SSA, anti-La/SSB, and anti-nuclear antibodies. [32][33]. In vitro findings showed that the transfection of LAMP3 expression plasmids in cultured SGECs triggered caspase-3 activity that leads to the apoptotic process [32][33]. Therefore, additional studies in vivo have demonstrated that the increased expression of LAMP3 provokes apoptosis in SGECs derived from non-obese diabetic (NOD) mice, a well-characterized model of the spontaneous onset of an SS-like phenotype, in which LAMP3 is locally overexpressed in the submandibular glands [33].
A schematic representation of the mechanisms described in the above paragraph is shown in Figure 2.
However, although many of the key apoptotic proteins that are activated or inactivated in apoptotic cascades have been discovered, the molecular events of the action or activation of these proteins in SS are not fully understood and are the focus of continued research.
3. Angiogenesis in SS
Angiogenesis is a fundamental process in growth, development, and repair [34]. Besides its well-known role in cancer, it has become clear that angiogenesis is also a critical component of non-neoplastic chronic inflammatory and autoimmune diseases, including atherosclerosis, RA, diabetic retinopathy, psoriasis, airway inflammation, peptic ulcers, Alzheimer’s disease, and SS [35][36]. In chronic inflammation, angiogenesis mediates the expansion of the microvascular tissue bed through the activation and proliferation of endothelial cells, leading to capillary and venule remodeling [37]. The expansion of the microvascular bed determines, in turn, the recruitment of inflammatory cells; for this reason, angiogenesis and inflammation seem to be chronically co-dependent processes [37].
3.1. Neo-Angiogenesis in pSS SGs
Neo-angiogenesis is mediated by the activity of vascular endothelial growth factor-A (VEGF-A) and its main receptor, vascular endothelial growth factor receptor-2 (VEGFR-2) [35][36][37][38]. In recent years it has emerged that, in pSS, infiltrating T cells and human SGECs produce increased amounts of pro-angiogenic factors via VEGF-A/VEGFR-2 system activation; consequently, VEGFR-2 blockade could be an entirely novel approach to blocking experimental angiogenesis and inflammation in pSS [39][40][41]. This agrees with the observation that the physiological response to tissue injury or infection is an increase in vascular permeability, and the microvascular changes associated with angiogenesis are key contributors to the tissue damage and remodeling processes that inevitably accompany chronic inflammation [39][40][41]. However, to date, few data are available on the role of angiogenesis in SS. Published data demonstrate that a functional impairment of the arterial wall may sustain early phases of atherosclerotic damage in pSS, and correlated chronic inflammation and immunological factors appear to be involved in the dysfunction of endothelial and vascular smooth muscle cells [40][41]. Confirming these hypotheses, strong positive staining for VEGF-A and VEGFR-2 proteins in pSS SG biopsies was detected [38], and a great number of pro-angiogenic proteins are overproduced in pSS SGs [38][40]. Recent research has been enriched by the placement of neuropilin in this scenario, a transmembrane co-receptor for members of the VEGF family, first described as mediators of neuronal organization, which seems to promote angiogenesis in pSS through the activation of NF-κB. [42]. However, direct evidence of neo-angiogenesis in SS is still lacking, although the sprouting of new vessels from preexisting ones was detected, favoring active macrophage and histiocyte infiltration [41]. The vasculature of SGs plays a critical role in saliva secretion [41], and the analysis of the changes that occur in the blood vessels during the progression of SS could help the understanding of the hyposalivation symptom. By virtue of its great therapeutic potential, in recent years, further investigations have been carried out in this field, leading to discoveries that are not always concordant. McCall and colleagues [43] showed decreased VEGF expression levels in pSS SGs, along with similar blood and lymphatic vessel organization and volume fractions, suggesting that angiogenesis and lymphangiogenesis do not play a significant role in the progression of SS. These conclusions were made with the use of selective markers for endothelial cells.
3.2. Endothelial T Cells in pSS
On the contrary, experimental data collected by Alunno and colleagues demonstrated that endothelial dysfunctions were present in pSS patients, represented by abnormalities in the endothelial cell structure that allow favorable conditions for the formation of atherosclerotic plaques [44]. In physiological conditions, endothelial cells may be damaged by several stimuli, including shear stress and transmural pressure, but they are promptly replaced thanks to the release of endothelial progenitor cells (EPC) from the bone marrow, which migrate to the site of injury and undergo a full maturation process. The assessment of circulating EPC along with circulating endothelial microparticles (EMP), which act as surrogate biomarkers of endothelial dysfunction, allowed researchers to verify that this process occurs in SS [45]. In particular, an increase in EPC in parallel to an increase in EMP may suggest a compensatory mechanism to overcome endothelial cell damage [45].
Therefore, in recent years, another leading actor in the scenario of endothelial repair has been identified, so-called angiogenic T cells (Tang) characterized by the co-expression of CD3, CD31, and CXCR4 [46]. Although consistent endothelial damage is ongoing in pSS, as proven by an increased amount of circulating EMP compared to healthy controls, a counteracting mechanism leading to EPC release from the bone marrow also takes place [45]. The current observations that circulating Tang cells are also raised in pSS, that they are significantly correlated to their partner EPC, and that both Tang and EPC are significantly associated with the EULAR SS disease activity index (ESSDAI) unmask another facet of this complex process. However, this makes it even more difficult to understand why, although the endothelial repair machinery seems to be fully working, pSS patients still display higher cardiovascular risk, and those with higher disease activity are at even more risk than those with milder disease [46]. Furthermore, Tang cells, which are numerous and close to blood vessels in pSS MSG, can produce IL-17, a cytokine that was recently demonstrated to play a versatile role in the pathogenesis of SS [47]. A hypothetical scenario showing the recent advances in SS angiogenesis is reported in Figure 3.
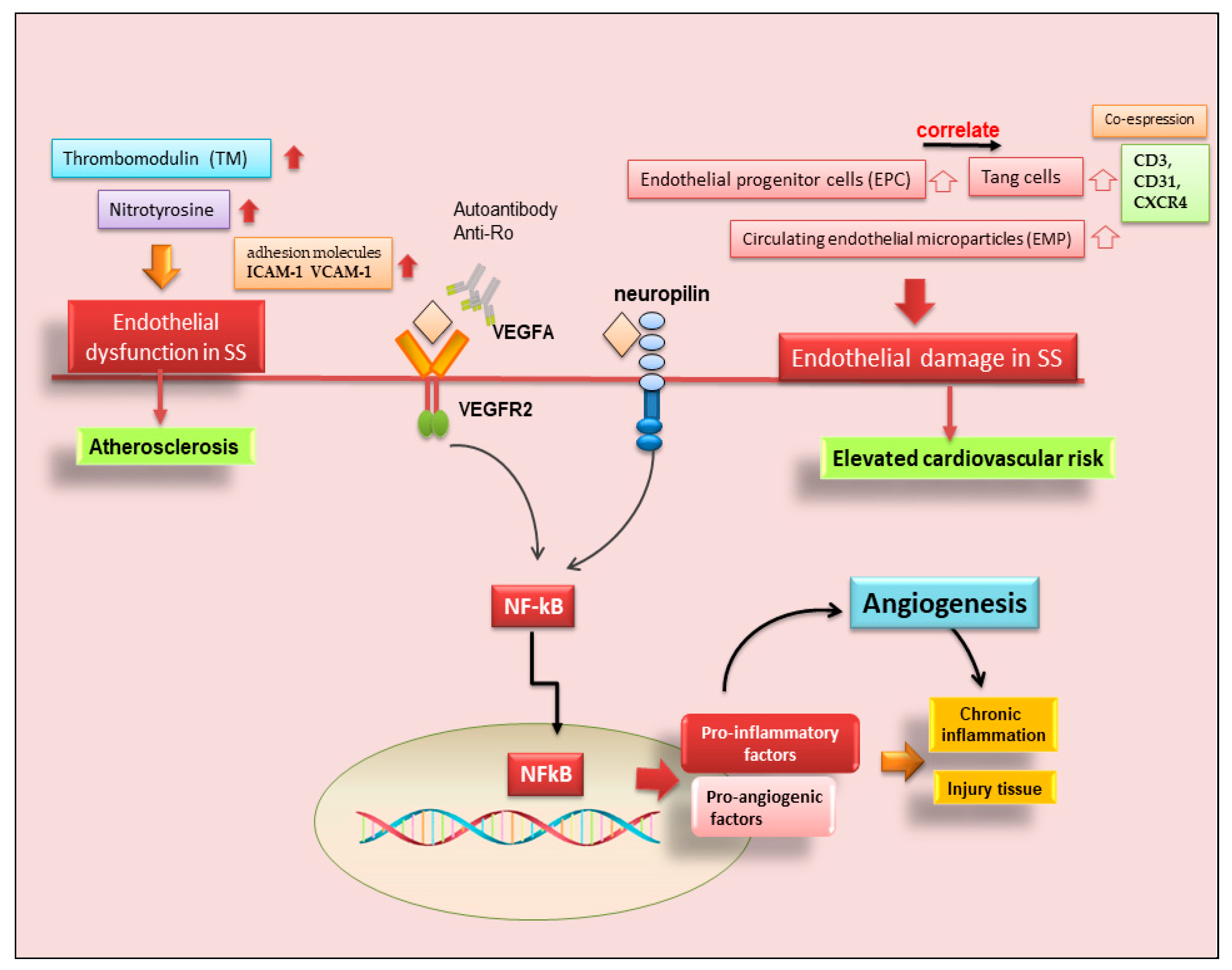
Figure 3. Role of angiogenesis in SS and SS-related diseases. Cluster of differentiation 3 (CD3); cluster of differentiation 31 (CD31); chemokine receptor type 4 (CXCR4); intercellular adhesion molecule 1 (ICAM-1); nuclear factor kappa B (NF-kB); vascular cell adhesion molecule 1 (VCAM-1); vascular endothelial growth factor A (VEGFA); vascular endothelial growth factor receptor 2 (VEGFR).
References
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165.
- Mariette, X.; Criswell, L.A. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018, 378, 931–939.
- Skarlis, C.; Raftopoulou, S.; Mavragani, C.P. Sjogren’s Syndrome: Recent Updates. J. Clin. Med. 2022, 11, 399.
- Wang, Y.; Xie, X.; Zhang, C.; Su, M.; Gao, S.; Wang, J.; Lu, C.; Lin, Q.; Lin, J.; Matucci-Cerinic, M.; et al. Rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren’s syndrome shared megakaryocyte expansion in peripheral blood. Ann. Rheum. Dis. 2022, 81, 379–385.
- Patel, Y.I.; McHugh, N.J. Apoptosis-new clues to the pathogenesis of Sjögren’s syndrome? Rheumatology 2000, 39, 119–121.
- Manganelli, P.; Fietta, P. Apoptosis and Sjögren syndrome. Semin. Arthritis Rheum. 2003, 33, 49–65.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Humphreys-Beher, M.G.; Peck, A.B.; Dang, H.; Talal, N. The role of apoptosis in the initiation of the autoimmune response in Sjögren’s syndrome. Clin. Exp. Immunol. 1999, 116, 383–387.
- Božič, B.; Rozman, B. Apoptosis and Autoimmunity. EJIFCC 2006, 17, 69–74.
- Ogawa, Y.; Takeuchi, T.; Tsubota, K. Autoimmune Epithelitis and Chronic Inflammation in Sjögren’s Syndrome-Related Dry Eye Disease. Int. J. Mol. Sci. 2021, 22, 11820.
- Shibata, Y.; Hishikawa, Y.; Izumi, S.; Fujita, S.; Yamaguchi, A.; Koji, T. Involvement of Fas/Fas ligand in the induction of apoptosis in chronic sialadenitis of minor salivary glands including Sjögren’s syndrome. Hum. Cell 2002, 15, 52–60.
- Nakamura, H.; Kawakami, A.; Izumi, M.; Nakashima, T.; Takagi, Y.; Ida, H.; Nakamura, T.; Nakamura, T.; Eguchi, K. Detection of the soluble form of Fas ligand (sFasL) and sFas in the saliva from patients with Sjögren’s syndrome. Clin. Exp. Rheumatol. 2005, 23, 915.
- Nakamura, H.; Horai, Y.; Shimizu, T.; Kawakami, A. Modulation of Apoptosis by Cytotoxic Mediators and Cell-Survival Molecules in Sjögren’s Syndrome. Int. J. Mol. Sci. 2018, 19, 2369.
- Haneji, N.; Nakamura, T.; Takio, K.; Yanagi, K.; Higashiyama, H.; Saito, I.; Noji, S.; Sugino, H.; Hayashi, Y. Identification of α-fodrin as a candidate autoantigen in primary Sjögren’s syndrome. Science 1997, 276, 604–607.
- Bulosan, M.; Pauley, K.M.; Yo, K.; Chan, E.K.; Katz, J.; Peck, A.B.; Cha, S. Inflammatory caspases are critical for enhanced cell death in the target tissue of Sjögren’s syndrome before disease onset. Immunol. Cell Biol. 2009, 87, 81–90.
- Casiano, C.A.; Martin, S.J.; Green, D.R.; Tan, E.M. Selective cleavage of nuclear autoantigens during CD95 (Fas/Apo-1)-mediated T cell apoptosis. J. Exp. Med. 1996, 184, 765–770.
- Sandhya, P.; Kurien, B.T.; Danda, D.; Scofield, R.H. Update on Pathogenesis of Sjogren’s Syndrome. Curr. Rheumatol. Rev. 2017, 13, 5–22.
- Ramos-Casals, M.; Font, J. Primary Sjögren’s syndrome: Current and emergent aetiopathogenic concepts. Rheumatology 2005, 44, 1354–1367.
- Sisto, M.; Lisi, S.; Castellana, D.; Scagliusi, P.; D’Amore, M.; Caprio, S.; Scagliusi, A.; Acquafredda, A.; Panaro, M.A.; Mitolo, V. Autoantibodies from Sjögren’s syndrome induce activation of both the intrinsic and extrinsic apoptotic pathways in human salivary gland cell line A-253. J. Autoimmun. 2006, 27, 38–49.
- Deets, K.A.; Vance, R.E. Inflammasomes and Adaptive Immune Responses. Nat. Immunol. 2021, 22, 412–422.
- Hachim, M.Y.; Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Pyroptosis: The Missing Puzzle Among Innate and Adaptive Immunity Crosstalk. J. Leukoc. Biol. 2020, 108, 323–338.
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging Insights into Molecular Mechanisms Underlying Pyroptosis and Functions of Inflammasomes in Diseases. J. Cell Physiol. 2020, 235, 3207–3221.
- Vakrakou, A.G.; Boiu, S.; Ziakas, P.D.; Xingi, E.; Boleti, H.; Manoussakis, M.N. Systemic Activation of NLRP3 Inflammasome in Patients with Severe Primary Sjögren’s Syndrome Fueled by Inflammagenic DNA Accumulations. J. Autoimmun. 2018, 91, 23–33.
- Vakrakou, A.G.; Svolaki, I.P.; Evangelou, K.; Gorgoulis, V.G.; Manoussakis, M.N. Cell-Autonomous Epithelial Activation of AIM2 (Absent in Melanoma-2) Inflammasome by Cytoplasmic DNA Accumulations in Primary Sjögren’s Syndrome. J. Autoimmun. 2020, 108, 102381.
- Hong, S.M.; Lee, J.; Jang, S.G.; Lee, J.; Cho, M.L.; Kwok, S.K.; Park, S.H. Type I Interferon Increases Inflammasomes Associated Pyroptosis in the Salivary Glands of Patients with Primary Sjögren’s Syndrome. Immune. Netw. 2020, 20, e39.
- Church, J.A. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Pediatrics 2014, 34, S184.
- Liu, Z.; Chu, A. Sjögren’s Syndrome and Viral Infections. Rheumatol. Ther. 2021, 8, 1051–1059.
- Nakamura, H.; Shimizu, T.; Kawakami, A. Role of Viral Infections in the Pathogenesis of Sjögren’s Syndrome: Different Characteristics of Epstein-Barr Virus and HTLV-1. J. Clin. Med. 2020, 9, 1459.
- Ishimaru, N.; Arakaki, R.; Omotehara, F.; Yamada, K.; Mishima, K.; Saito, I.; Hayashi, Y. Novel role for RbAp48 in tissue-specific, estrogen deficiency-dependent apoptosis in the exocrine glands. Mol. Cell. Biol. 2006, 26, 2924–2935.
- Ishimaru, N.; Arakaki, R.; Yoshida, S.; Yamada, A.; Noji, S.; Hayashi, Y. Expression of the retinoblastoma protein RbAp48 in exocrine glands leads to Sjögren’s syndrome-like autoimmune exocrinopathy. J. Exp. Med. 2008, 205, 2915–2927.
- Otsuka, K.; Sato, M.; Tsunematsu, T.; Ishimaru, N. Virus Infections Play Crucial Roles in the Pathogenesis of Sjögren’s Syndrome. Viruses 2022, 14, 1474.
- Tanaka, T.; Warner, B.M.; Odani, T.; Ji, Y.; Mo, Y.Q.; Nakamura, H.; Jang, S.I.; Yin, H.; Michael, D.G.; Hirata, N.; et al. LAMP3 induces apoptosis and autoantigenrelease in Sjögren’s syndrome patients. Sci. Rep. 2020, 10, 15169.
- Nakamura, H.; Tanaka, T.; Pranzatelli, T.; Ji, Y.; Yin, H.; Perez, P.; Afione, S.A.; Jang, S.I.; Goldsmith, C.; Zheng, C.Y.; et al. Lysosome-associated membrane protein 3 misexpression in salivary glands induces a Sjögren’s syndrome-like phenotype in mice. Ann. Rheum. Dis. 2021, 80, 1031–1039.
- Ribatti, D.; Crivellato, E. Sprouting angiogenesis, a reappraisal. Dev. Biol. 2012, 15, 157–165.
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936.
- Majno, G. Chronic inflammation: Links with angiogenesis and wound healing. Am. J. Pathol. 1998, 153, 1035–1039.
- Bagli, E.; Xagorari, A.; Papetropoulos, A.; Murphy, C.; Fotsis, T. Angiogenesis in inflammation. Autoimmun. Rev. 2004, 3, S26.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Frassanito, M.A.; Ribatti, D. Sjögren’s syndrome pathological neovascularization is regulated by VEGF-A-stimulated TACE-dependent crosstalk between VEGFR2 and NF-κB. Genes Immun. 2012, 13, 411–420.
- Abu-Helu, R.F.; Dimitriou, I.D.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Manoussakis, M.N. Induction of salivary gland epithelial cell injury in Sjogren’s syndrome: In vitro assessment of T cell-derived cytokines and Fas protein expression. J. Autoimmun. 2001, 17, 141–153.
- Gerli, R.; Vaudo, G.; Bocci, E.B. Functional impairment of the arterial wall in primary Sjögren’s syndrome: Combined action of immunologic and inflammatory factors. Arthritis Care Res. 2010, 62, 712–718.
- Thakor, A.S.; Brown, C.N.; Edwards, A. Effects of prolonged reduction in blood flow on submandibular secretory function in anesthetized sheep. J. Appl. Physiol. 2003, 95, 751–757.
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Ribatti, D. Neuropilin-1 is upregulated in Sjögren’s syndrome and contributes to pathological neovascularization. Histochem. Cell Biol. 2012, 137, 669–677.
- McCall, A.D.; Baker, O.J. Characterization of Angiogenesis and Lymphangiogenesis in Human Minor Salivary Glands with Sjögren’s Syndrome. J. Histochem. Cytochem. 2015, 63, 340–349.
- Alunno, A.; Ibba-Manneschi, L.; Bistoni, O.; Cipriani, S.; Topini, F.; Gerli, R.; Manetti, M. Angiogenic T cells in primary Sjögren’s syndrome: A double-edged sword? Clin. Exp. Rheumatol. 2019, 118, 36–41.
- Bartoloni, E.; Alunno, A.; Bistoni, O.; Caterbi, S.; Luccioli, F.; Santoboni, G.; Mirabelli, G.; Cannarile, F.; Gerli, R. Characterization of circulating endothelial microparticles and endothelial progenitor cells in primary Sjögren’s syndrome: New markers of chronic endothelial damage? Rheumatology 2015, 54, 536–544.
- Hur, J.; Yang, H.M.; Yoon, C.H.; Lee, C.S.; Park, K.W.; Kim, J.H.; Kim, T.Y.; Kim, J.Y.; Kang, H.J.; Chae, I.H.; et al. Identification of a novel role of T cells in postnatal vasculogenesis: Characterization of endothelial progenitor cell colonies. Circulation 2007, 116, 1671–1682.
- Sisto, M.; Lorusso, L.; Tamma, R.; Ingravallo, G.; Ribatti, D.; Lisi, S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren’s syndrome. Clin. Exp. Immunol. 2019, 198, 261–272.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.7K
Revisions:
2 times
(View History)
Update Date:
01 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No