 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vinicius Cruzat | + 2889 word(s) | 2889 | 2020-11-16 03:50:44 | | | |
| 2 | Bruce Ren | Meta information modification | 2889 | 2020-11-25 06:36:08 | | | | |
| 3 | Bruce Ren | Meta information modification | 2889 | 2021-01-27 10:16:27 | | |
Video Upload Options
Type 2 diabetes (T2D) and Alzheimer’s disease (AD) are growing in prevalence worldwide. The development of T2D increases the risk of AD disease, while AD patients can show glucose imbalance due to an increased insulin resistance. T2D and AD share similar pathological features and underlying mechanisms, including the deposition of amyloidogenic peptides in pancreatic islets (i.e., islet amyloid polypeptide; IAPP) and brain (β-Amyloid; Aβ). Both IAPP and Aβ can undergo misfolding and aggregation and accumulate in the extracellular space of their respective tissues of origin. As a main response to protein misfolding, there is evidence of the role of heat shock proteins (HSPs) in moderating T2D and AD. HSPs play a pivotal role in cell homeostasis by providing cytoprotection during acute and chronic metabolic stresses. In T2D and AD, intracellular HSP (iHSP) levels are reduced, potentially due to the ability of the cell to export HSPs to the extracellular space (eHSP). The increase in eHSPs can contribute to oxidative damage and is associated with various pro-inflammatory pathways in T2D and AD.
1. Introduction
Alzheimer’s disease (AD) and type 2 diabetes (T2D) are two of the most widespread age-related chronic diseases, and the prevalence of both is steadily increasing [1][2][3]. AD and T2D share similar risk factors, which can include a sedentary lifestyle, poor diet, obesity, and hereditary predisposition [3]. Studies have shown that patients with T2D are up to 65% more likely to develop AD than non-diabetic patients, while AD individuals are more likely to be insulin resistant [4][5]. T2D and AD also share dysfunctions in the insulin receptor, chronic inflammation, and secretion of amyloidogenic peptides [6][7].
Amyloidogenic peptides are peptides that spontaneously misfold, aggregate, and deposit in extracellular spaces, forming toxic soluble intermediates and insoluble fibrillar amyloid plaque. Amyloidogenic peptides are associated with the development of T2D and AD through the formation of islet amyloid polypeptide (IAPP) and β-amyloid (Aβ), respectively [8][9][10]. These two amyloidogenic peptides have similar methods of exerting toxicity involving membrane pore formation, mitochondrial dysfunction, oxidative stress, endoplasmic reticulum (ER) stress, and apoptosis [11][12]. While Aβ and IAPP deposit in their respective tissues of origin, they also co-localize in the plaque of both brain tissue and pancreatic islets, where they undergo misfolding and aggregation [13][14]. Once co-localized, Aβ and IAPP may also undergo a process called cross-seeding, where the aggregation and seeding of amyloidogenic peptides attract and aggregate with more similar and/or different types of amyloidogenic peptides [15][16]. In this case, co-localized Aβ and IAPP may promote the formation of combined Aβ-IAPP oligomeric hetero-complexes [17][18][19]. Such dysfunctions in the protein homeostasis of vital tissues often create a stressful environment in which cells may fail to thrive.
At the most basic cellular level, living organisms respond to stressful or unfavorable conditions by changing the expression of stress-related genes, predominantly via the transcription and upregulation of heat shock proteins (HSPs) [20]. This cytoprotective molecular organization is referred to as the heat shock response (HSR). HSPs are a class of proteins that are rapidly upregulated by cells in response to a variety of endogenous or exogenous stressors [20]. Despite being originally defined by their role in the thermal stress response [21], HSPs are now understood to be expressed both constitutively and in the presence of cellular stresses such as oxidative stress [22] and inflammation [22][23].
One of the main roles associated with the HSPs is to support protein maintenance, including assisting the proper folding of newly synthesized proteins, refolding and clearance of misfolded and/or aggregated proteins, and participating in the membrane translocation of secretory proteins [24]. As the folding, maintenance, and degradation of proteins are key requirements for cell homeostasis, alterations in the HSR and the associated functions of HSPs have been linked to chronic diseases, such as AD and T2D.
2. Heat Shock Response in Type 2 Diabetes and Alzheimer’s Disease
HSPs and the HSR have a predominantly cytoprotective role with the potential to attenuate T2D or AD pathology. HSPs can clear aggregated amyloid proteins and prevent further amyloid aggregation by inhibiting both the nucleation and elongation processes of cross-seeding [25][26]. However, both AD and T2D may feature an altered expression of HSPs, as the HSR is often dysregulated in aged and obese individuals, and phenotypes often seen in both of these two chronic metabolic-associated age-related diseases [27][28]. A downregulation of HSP and the HSR correlates with dysfunctional insulin signaling, which is another feature of both diseases, suggesting a strong correlation between HSPs and insulin signaling [29][30][31]. A potential reason for this involves glycogen synthase kinase-3 (GSK-3), which is a negative regulator of the insulin signaling cascade. An inflammatory environment resulting from chronic disease and obesity, such as in AD and T2D, negatively impact insulin signaling, which results in the activation of GSK-3 [31][32]. As well as further impairing insulin signaling, activated GSK-3 can phosphorylate HSF1 [33][34]. The phosphorylation of HSF1 inhibits its translocation to the nucleus, thereby lowering the gene expression of HSPs. The inhibition of GSK-3 causes an upregulation of HSP and restoration of insulin signaling [35][36]. Then, a vicious cycle can be established, where the inflammatory environment impairs insulin sensitivity and insulin signaling, which in turn impairs the cells’ ability to manage the stresses of the local environment via the downregulation of HSPs, making insulin-sensitive tissues more susceptible to damage and resulting in further increases in inflammation and hyperglycemia (Figure 1).
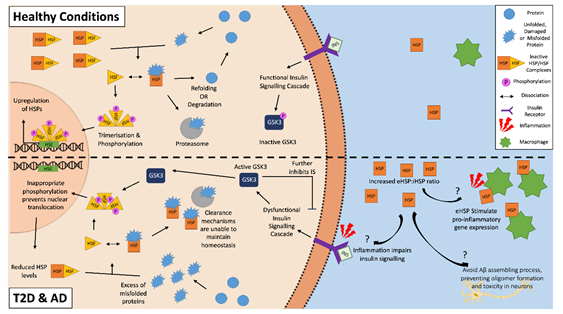
Figure 1. The heat shock response in Alzheimer’s disease and type 2 diabetes. The inflammatory environment in AD and T2D negatively disrupts insulin signaling, activating glycogen synthase kinase-3 (GSK-3), which in turn inappropriately phosphorylates heat shock factor 1 (HSF1). This phosphorylation inhibits the translocation of the HSF1 trimers to the nucleus, and as a result, inhibits the upregulation of HSPs. This reduced intracellular HSP pool is unable to effectively clear the aggregated amyloidogenic peptides within the cells. In the external environment, increased levels of extracellular HSPs found in T2D and AD patients can act as “chaperokines” and stimulate the immune system to produce pro-inflammatory factors. This becomes a vicious cycle of increased inflammation, decreased insulin signaling, and a decreased ability for HSP to clear aggregated peptides. On the other hand, some eHSPs (e.g., eHSP90 and eHSP70) in AD may have a role in the clearance of amyloid plaque, as they can modulate Aβ toxicity.
In neuronal tissue and pancreatic islets where amyloid plaque can accumulate, impaired insulin signaling can result in a low HSP environment, favoring an increased aggregation of amyloidogenic peptides. However, in a T2D monkey model, it was shown that despite decreases in HSP and HSF levels in peripheral tissues such as the liver, HSF1 expression was increased, and HSP levels were maintained in pancreatic tissue [37]. This could indicate a possible islet-specific mechanism for protection from the inflammatory environment compared to peripheral tissues at early stages of T2D.
It has also been suggested that decreased membrane integrity could contribute to the reduction of HSP and HSF levels. Membrane lipid defects affecting integrity, fluidity, and composition, are featured in both AD and T2D [38][39]. HSPs are known to mediate membrane integrity, supporting the cell during stressful conditions. Therefore, the combination of a reduction in HSPs and lipid defects induced by insulin insensitivity could further potentiate lipid membrane disruptions [40]. Considering that that there are several HSP proteins and that they have different locations and functions, the effects on the pathology in T2D and AD cannot be generalized amongst all members of the HSP/HSF family. Indeed, evidence to date implicate HSP90, 70, 60, and 40 in moderating Aβ, Tau, or IAPP aggregation, or cellular stress induced by these aggregated proteins [41]. There are also emerging roles for sHSP and extracellular eHSP in T2D and AD. These topics are discussed further in the present review.
2.1. Heat Shock Protein 90
HSP90 is the most abundantly expressed HSP protein in eukaryotic cells [42]. Predominantly located in the cytosol, the functions of HSP90s include mediating the inflammatory response, as well as stabilizing and correcting misfolded proteins . Experimental evidence suggests that HSP90s are involved in regulating the activity of several signaling proteins, such as steroid hormone receptors, and cellular differentiation processes.
Although the expression of HSP90 is predominantly constitutive, it can also be stress-induced in response to misfolded or aggregated proteins. HSP90 can inhibit Aβ toxicity by binding misfolded Aβ peptides [43]. Once bound, HSP90 then prevents further aggregation using an ATP-independent pathway or by changing the conformation of Aβ to a state less prone to aggregation via an ATP-dependent pathway [43]. In addition, tau is a substrate protein for HSP90 chaperones, where they can bind hyperphosphorylated tau and activate degradation processes [44][45]. As for IAPP, evidence shows that the ubiquitin–proteosomal system, which includes HSP90, is important for IAPP clearance and turnover. A decline in the function of this system due to inflammation and aging is detrimental to pancreatic islets, allowing IAPP aggregation to occur [46][47][48].
The role of HSP90 in T2D and AD is complex, as HSP90 can reportedly have both protective and detrimental roles in managing amyloidogenic peptides. The inhibition of HSP90 promoted an increased clearance of Aβ and tau in primary neuronal cells of rats [49][50], and it also facilitated better glucose regulation in T2D mice [51]. HSP90 inhibition also increased the HSR by increasing the dissociation of HSF1 from HSP90, where it could translocate to the nucleus and activate/potentiate the HSR [49]. HSP90 assists in the maintenance and function of GSK-3 [50], which is well known for its detrimental effects in T2D and AD [81,84]. In addition, HSP90 may even facilitate tau aggregation and hyperphosphorylation in rat brain extracts by promoting conformation changes in tau that promote its phosphorylation by GSK-3[52].
As HSP90 actions can vary so widely, post-translational modifications and co-chaperones are among the most important factors in the regulation of HSP90 activity. This includes the acetylation and/or phosphorylation of HSP90, as well as the formation of larger protein complexes with other HSPs, particularly HSP70 and HSP40 [53]. The HSP90/70/40 complex can slow Aβ aggregation in a chaperone dose-dependent manner. In addition, in brain tissue from both transgenic AD mouse models and AD patients, expression or formation of the HSP90/70/40 complex has been found to inversely correlate with tau aggregation [54], supporting a protective role for this complex.
2.2. Heat Shock Protein 70
The HSP70 family is a varied group of chaperones with wide-ranging functions and subcellular locations. A defining feature of the group, aside from their similar molecular weight, is the shared structure of a substrate binding domain at the C-terminus that bind polypeptides, and at the N-terminus, a nucleotide binding domain (NBD) that interacts with ATPase to hydrolyze ATP [55]. Members of this family include the constitutively expressed Heat Shock Cognate 70 (HSC70) and the stress-induced HSP72, which are both found in the cytosol. Other members of the HSP70 family are found in organelle-specific locations, such as glucose-responsive proteins 78 (GRP78) and 75 (GRP75) are localized in the ER and mitochondria, respectively [56].
HSC70, as a constitutive HSP, is involved in general proteostatic functions, such as the support of protein assembly and protein trafficking throughout the cell. It also has roles in the innate immune response, as well as cell differentiation processes [57]. However, during cellular stress, HSP72 is one of the most strongly induced chaperones in the HSR and is considered one of the main stress-responsive chaperones in cells [58].
In T2D patients, HSP72 has been reported to inhibit the aggregation of IAPP [59][60]. Heat therapy to induce HSP72 was found to reduce insulin resistance and improve clinical parameters in T2D patients. This was suspected to result from an HSP72-induced reduction of pro-inflammatory signaling molecule phosphorylation, which impaired normal insulin responses. HSP72 can increase the fatty acid oxidation capacity in skeletal muscle, protecting against increases in insulin resistance and body weight [60]. Furthermore, HSP72 mRNA expression in skeletal muscle correlates with mitochondrial enzyme activity, rate of lipid turnover, and insulin-stimulated glucose uptake [61]. However, skeletal muscle HSP72 mRNA expression has been shown to be reduced in T2D patients compared to controls, suggesting a less efficient HSR . In addition, HSP70 can be susceptible to glycation in a hyperglycemic environment, further reducing its chaperone activity [62].
In AD, the HSP70 proteins have cytoprotective roles via different mechanisms, including the inhibition of Aβ oligomerisation and remodeling to a less amyloidogenic form [44], upregulation of Aβ degradation enzymes [63][64], and restoring tau homeostasis by promoting the degradation of pTau aggregates, most likely by the ubiquitin–proteasome and/or autophagy systems [64]. However, HSC70 and HSP72 had opposite effects on tau stability; HSP72 increases the degradation of tau, while HSC70 reduces it. The ratio of inducible to constitutive HSPs appears to be a critical factor, as increased levels of HSP72 appears to negate the ability of HSC70 to stabilize Tau [65][66].
Other members of the HSP70 family, such as GRP78, also have documented roles in AD pathology. Intracellular Aβ oligomers can cause cellular damage before becoming extracellular. To prevent Aβ intracellular toxicity, GRP78 can bind to Aβ precursor proteins in the ER, preventing the β/γ-secretase cleavage necessary to process APP to Aβ, as shown in an HEK cell model co-transfected with APP and GRP78 [67].
2.3. Heat Shock Protein 60
HSP60 chaperones are traditionally associated with the mitochondria, and various studies have indicated that HSP60 may be more ubiquitously expressed than previously thought [68][69]. Within the mitochondria, HSP60 works closely with co-factor HSP10 (a member of the sHSP family) to maintain and correct the folding of mitochondrial proteins. If a deficiency in these HSPs were to occur, as observed in skeletal muscle of T2D patients [70] and the cortex of AD patients [71], cellular stress is enhanced.
Neuroprotective effects of HSP60 have been demonstrated in a human neuroblastoma cell line, where the overexpression of HSP60 inhibited an Aβ-induced reduction of Cytochrome C Oxidase (COX) IV activity in the mitochondria, subsequently reducing apoptosis [72]. However, HSP60 has also been implicated in pro-apoptotic functions in cells. HSP60 was shown to bind to pro-caspase 3 in vitro and accelerate its maturation during apoptosis [73][74]. Furthermore, HSP60 can mediate the mislocalisation of APP to the mitochondria, where Aβ peptides can aggregate, possibly leading to mitochondrial dysfunction [75]. Considering these somewhat opposite effects reported for HSP60 on AD pathology, further investigation is warranted, potentially examining the role of expression or interactions with other HSPs that moderate the overall activities of HSP60.
2.4. Heat Shock Protein 40
The HSP40 family of chaperones function differently compared to other HSPs, as they require co-chaperones from the HSP70 group to be active [76]. HSP40 uses a conserved N-Terminus J-Domain to bind to the ATPase N-Terminus of HSP70s, stimulating ATP’s conversion to adenosine diphosphate (ADP) via hydrolysis. Once this occurs, HSP70 becomes activated, dissociates from HSF1, and begins binding to non-native proteins [77]. HSP40 may also bind to substrates and escort them to the substrate-binding domain of HSP70, where they mediate the process of refolding the substrate proteins. This HSP70/40 complex greatly enhances the efficiency and capability of the refolding cycle, including increasing ATP hydrolysis rate up to 1000-fold over basal levels [78].
While HSP40 is not directly involved in the pathogenesis of T2D or AD, it can facilitate and regulate many of the HSPs that are—for example, the HSP70/40 complex, which has been shown to inhibit Aβ aggregation in neuronal cells. Interestingly, the B3 member of the HSP40 co-chaperone family, along with HSP72, has been shown to mediate glucose uptake and insulin signaling via c-Jun N-terminal kinase (JNK) repression [79]. Thus, alterations in the expression or function of HSPs in tissues associated with amyloidogenic peptides could result in the promotion of chronic disease such as T2D and AD.
2.5. Small Heat Shock Proteins
sHSPs are a family of ATP-independent chaperones with sizes ranging from 10 to 40 kDa that share common features, including a conserved α-crystallin domain. This domain allows for dimerization, leading to oligomeric assembly of the sHSPs, where they can then successfully bind non-native protein substrates and form stable sHSP–substrate complexes. The oligomeric complexes are known for binding to several non-native proteins at once, which is a feature that is absent in other molecular chaperone families [80][81]. ATP-dependent chaperones are required to release the non-native protein from the sHSP–substrate complex, where it can then be refolded or degraded. By creating a reservoir of proteins for refolding or degradation, the presence of sHSPs makes the process more efficient.
sHSP have vital roles in T2D and AD. The expression of sHSPs is upregulated in T2D-associated tissues, including skeletal muscle, retina, and cardiac muscle. However, their ability to function correctly is often negatively affected, as both the solubility and activation of specific sHSPs is reduced in hyperglycemic environments [82][83]. In AD, the HSP27 species of sHSP preferentially interacts with hyperphosphorylated tau in human brain samples. In vitro, HSP27 modulates tau pathology by decreasing the level of hyperphosphorylated tau, suppressing tau-induced apoptosis and increasing the amount of dephosphorylated tau [84]. Furthermore, sHSP species can directly bind to Aβ and IAPP to prevent fibril formation [85] and aggregation. Aβ binding to metal ions, such as Cu2+, forms metal–peroxidases, which contribute to oxidative stress and an increased aggregation of Aβ. sHSP chaperones have been shown to prevent the Cu2+-induced aggregation by dislodging the bound Cu2+ ion, preventing fibril formation [86]. However, preventing fibril formation may not result in reductions in amyloid-induced toxicity. When the sHSP, αB-crystallin, was co-incubated with Aβ peptides in vitro, it inhibited the aggregation of Aβ into fibrils and instead maintained the Aβ peptides in oligomeric αB-crystallin/Aβ complexes that were more toxic to neuronal cells [87].
References
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053.
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117, doi:10.1016/s0140-6736(05)67889-0.
- Akter, K.; Lanza, E.A.; Martin, S.A.; Myronyuk, N.; Rua, M.; Raffa, R.B. Diabetes mellitus and Alzheimer’s disease: Shared pathology and treatment? Br. J. Clin. Pharmacol. 2011, 71, 365–376, doi:10.1111/j.1365-2125.2010.03830.x.
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666, doi:10.1001/archneur.61.5.661.
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481.
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272, doi:https://doi.org/10.1016/j.ceca.2009.12.010.
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635, doi:10.1007/s00125-008-1255-x.
- Cohen, A.S.; Calkins, E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 1959, 183, 1202.
- Epstein, F.H.; Höppener, J.W.M.; Ahrén, B.; Lips, C.J.M. Islet Amyloid and Type 2 Diabetes Mellitus. N. Engl. J. Med. 2000, 343, 411–419, doi:10.1056/NEJM200008103430607.
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24, doi:10.1038/sj.cdd.4401528.
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675, doi:10.1073/pnas.0903563106.
- Mirzabekov, T.A.; Lin, M.C.; Kagan, B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996, 271, 1988–1992.
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Laszlo, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol. Aging 2010, 31, 1503–1515, doi:10.1016/j.neurobiolaging.2008.08.019.
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526, doi:10.1002/ana.23956.
- Berhanu, W.M.; Yaşar, F.; Hansmann, U.H.E. In Silico Cross Seeding of Aβ and Amylin Fibril-like Oligomers. ACS Chem. Neurosci. 2013, 4, 1488–1500, doi:10.1021/cn400141x.
- Baram, M.; Atsmon-Raz, Y.; Ma, B.; Nussinov, R.; Miller, Y. Amylin-Abeta oligomers at atomic resolution using molecular dynamics simulations: A link between Type 2 diabetes and Alzheimer’s disease. Phys. Chem. Chem. Phys. Pccp 2016, 18, 2330–2338, doi:10.1039/c5cp03338a.
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356, doi:10.1038/s41598-020-66602-9.
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. JAD 2017, 59, 421–432, doi:10.3233/jad-161192.
- Martinez-Valbuena, I.; Valenti-Azcarate, R.; Amat-Villegas, I.; Riverol, M.; Marcilla, I.; de Andrea, C.E.; Sánchez-Arias, J.A.; Del Mar Carmona-Abellan, M.; Marti, G.; Erro, M.E.; et al. Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann. Neurol. 2019, 86, 539–551, doi:10.1002/ana.25570.
- Kampinga, H.; Hageman, J.; Vos, M.; Kubota, H.; Tanguay, R.; Bruford, E.; Cheetham, M.; Chen, B.; Hightower, L. Guidelines for the nomenclature of the human heat shock proteins. A Compr. J. Stress Biol. Med. 2009, 14, 105–111, doi:10.1007/s12192-008-0068-7.
- Yang, X.M.; Baxter, G.F.; Heads, R.J.; Yellon, D.M.; Downey, J.M.; Cohen, M.V. Infarct limitation of the second window of protection in a conscious rabbit model. Cardiovasc. Res. 1996, 31, 777–783, doi:10.1016/0008-6363(96)00026-0.
- Krause, M.S.; Oliveira, L.P.; Silveira, E.M.S.; Vianna, D.R.; Rossato, J.S.; Almeida, B.S.; Rodrigues, M.F.; Fernandes, A.J.M.; Costa, J.A.B.; Curi, R.; et al. MRP1/GS-X pump ATPase expression: Is this the explanation for the cytoprotection of the heart against oxidative stress-induced redox imbalance in comparison to skeletal muscle cells? Cell Biochem. Funct. 2007, 25, 23, doi:10.1002/cbf.1343.
- Cruzat, V.F.; Pantaleao, L.C.; Donato, J., Jr.; de Bittencourt, P.I., Jr.; Tirapegui, J. Oral supplementations with free and dipeptide forms of L-glutamine in endotoxemic mice: Effects on muscle glutamine-glutathione axis and heat shock proteins. J. Nutr. Biochem. 2014, 25, 345–352, doi:10.1016/j.jnutbio.2013.11.009.
- Edkins, A.L.; Price, J.T.; Pockley, A.G.; Blatch, G.L. Heat shock proteins as modulators and therapeutic targets of chronic disease: An integrated perspective. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2018, 373, 20160521, doi:10.1098/rstb.2016.0521.
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012, 10, e1001346, doi:10.1371/journal.pbio.1001346.
- Arimon, M.; Grimminger, V.; Sanz, F.; Lashuel, H.A. Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Abeta fibrils and protofibrils. J. Mol. Biol. 2008, 384, 1157–1173, doi:10.1016/j.jmb.2008.09.063.
- Tower, J. Hsps and aging. Trends Endocrinol. Metab. 2009, 20, 216–222, doi:10.1016/j.tem.2008.12.005.
- Sabbah, N.A.; Rezk, N.A.; Saad, M.S.S. Relationship Between Heat Shock Protein Expression and Obesity With and Without Metabolic Syndrome. Genet. Test. Mol. Biomark. 2019, 23, 737–743, doi:10.1089/gtmb.2019.0062.
- Kurucz, I.; Morva, A.; Vaag, A.; Eriksson, K.F.; Huang, X.; Groop, L.; Koranyi, L. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes 2002, 51, 1102–1109.
- Vigh, L.; Horvath, I.; Maresca, B.; Harwood, J.L. Can the stress protein response be controlled by ‘membrane-lipid therapy’? Trends Biochem. Sci. 2007, 32, 357–363, doi:10.1016/j.tibs.2007.06.009.
- Hooper, P.L.; Hooper, P.L. Inflammation, heat shock proteins, and type 2 diabetes. Cell Stress Chaperones 2009, 14, 113–115, doi:10.1007/s12192-008-0073-x.
- Patel, S.; Doble, B.; Woodgett, J.R. Glycogen synthase kinase-3 in insulin and Wnt signalling: A double-edged sword? Biochem. Soc. Trans. 2004, 32, 803–808, doi:10.1042/bst0320803.
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441.
- Kurthy, M.; Mogyorosi, T.; Nagy, K.; Kukorelli, T.; Jednakovits, A.; Talosi, L.; Biro, K. Effect of BRX-220 against peripheral neuropathy and insulin resistance in diabetic rat models. Ann. N. Y. Acad. Sci. 2002, 967, 482–489.
- Hooper, P.L. Insulin Signaling, GSK-3, Heat Shock Proteins and the Natural History of Type 2 Diabetes Mellitus: A Hypothesis. Metab. Syndr. Relat. Disord. 2007, 5, 220–230, doi:10.1089/met.2007.0005.
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439, doi:10.1111/j.1471-4159.2007.05194.x.
- Kavanagh, K.; Zhang, L.; Wagner, J.D. Tissue-specific regulation and expression of heat shock proteins in type 2 diabetic monkeys. Cell Stress Chaperones 2009, 14, 291–299, doi:10.1007/s12192-008-0084-7.
- Ginsberg, L.; Atack, J.R.; Rapoport, S.I.; Gershfeld, N.L. Evidence for a membrane lipid defect in Alzheimer disease. Mol. Chem. Neuropathol. 1993, 19, 37–46, doi:10.1007/BF03160167.
- Weijers, R.N.M. Lipid Composition of Cell Membranes and Its Relevance in Type 2 Diabetes Mellitus. Curr. Diabetes Rev. 2012, 8, 390–400, doi:10.2174/157339912802083531.
- Horváth, I.; Multhoff, G.; Sonnleitner, A.; Vígh, L. Membrane-associated stress proteins: More than simply chaperones. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 1653–1664, doi:10.1016/j.bbamem.2008.02.012.
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11, doi:10.1038/nrn1587.
- Mickler, M.; Hessling, M.; Ratzke, C.; Buchner, J.; Hugel, T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Amp; Mol. Biol. 2009, 16, 281, doi:10.1038/nsmb.1557.
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat Shock Proteins 70 and 90 Inhibit Early Stages of Amyloid β-(1–42) Aggregation in Vitro. J. Biol. Chem. 2006, 281, 33182–33191, doi:10.1074/jbc.M606192200.
- Dickey, C.A.; Yue, M.; Lin, W.-L.; Dickson, D.W.; Dunmore, J.H.; Lee, W.C.; Zehr, C.; West, G.; Cao, S.; Clark, A.M.K.; et al. Deletion of the Ubiquitin Ligase CHIP Leads to the Accumulation, But Not the Aggregation, of Both Endogenous Phospho- and Caspase-3-Cleaved Tau Species. J. Neurosci. 2006, 26, 6985–6996, doi:10.1523/jneurosci.0746-06.2006.
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263, doi:10.1111/j.1471-4159.2005.03272.x.
- Press, M.; Jung, T.; König, J.; Grune, T.; Höhn, A. Protein aggregates and proteostasis in aging: Amylin and β-cell function. Mech. Ageing Dev. 2019, 177, 46–54, doi:https://doi.org/10.1016/j.mad.2018.03.010.
- Chatterjee Bhowmick, D.; Jeremic, A. Functional proteasome complex is required for turnover of islet amyloid polypeptide in pancreatic β-cells. J. Biol. Chem. 2018, 293, 14210–14223, doi:10.1074/jbc.RA118.002414.
- Singh, S.; Trikha, S.; Sarkar, A.; Jeremic, A.M. Proteasome regulates turnover of toxic human amylin in pancreatic cells. Biochem. J. 2016, 473, 2655–2670, doi:10.1042/BCJ20160026.
- Zhao, H.; Michaelis, M.L.; Blagg, B.S.J. Hsp90 Modulation for the Treatment of Alzheimer’s Disease. In Advances in Pharmacology; Michaelis, E.K., Michaelis, M.L., Eds.; Academic Press: 2012; Cambridge, USA Volume 64, pp. 1–25.
- Dou, F.; Chang, X.; Ma, D. Hsp90 Maintains the Stability and Function of the Tau Phosphorylating Kinase GSK3β. Int. J. Mol. Sci. 2007, 8, 51–60.
- Lee, J.-H.; Gao, J.; Kosinski, P.A.; Elliman, S.J.; Hughes, T.E.; Gromada, J.; Kemp, D.M. Heat shock protein 90 (HSP90) inhibitors activate the heat shock factor 1 (HSF1) stress response pathway and improve glucose regulation in diabetic mice. Biochem. Biophys. Res. Commun. 2013, 430, 1109–1113, doi:https://doi.org/10.1016/j.bbrc.2012.12.029.
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. JAD 2009, 17, 319–325, doi:10.3233/jad-2009-1049.
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 Chaperone Machinery. J. Biol. Chem. 2008, 283, 18473–18477, doi:10.1074/jbc.R800007200.
- Ou, J.-R.; Tan, M.-S.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat Shock Protein 90 in Alzheimer’s Disease. Biomed Res. Int. 2014, 2014, 796869, doi:10.1155/2014/796869.
- Young, J.C. Mechanisms of the Hsp70 chaperone system. Biochem. Cell Biol. Biochim. Biol. Cell. 2010, 88, 291–300, doi:10.1139/o09-175.
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710, doi:https://doi.org/10.1016/j.febslet.2007.05.039.
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sorensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105, doi:10.1038/nature05378.
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell. Mol. Med. 2008, 12, 743–761, doi:10.1111/j.1582-4934.2008.00273.x.
- Chilukoti, N.; Sahoo, B.; Maddheshiya, M.; Garai, K. Hsp70 Delays Amyloid Aggregation of Amylin by Inhibiting Primary Nucleation. Biophys. J. 2018, 114, 78a–79a, doi:10.1016/j.bpj.2017.11.475.
- Chung, J.; Nguyen, A.-K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.S.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744, doi:10.1073/pnas.0705799105.
- Henstridge, D.C.; Whitham, M.; Febbraio, M.A. Chaperoning to the metabolic party: The emerging therapeutic role of heat-shock proteins in obesity and type 2 diabetes. Mol. Metab. 2014, 3, 781–793, doi:10.1016/j.molmet.2014.08.003.
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C.; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459, doi:10.1096/fj.12-220889.
- Jinwal, U.K.; Miyata, Y.; Koren, J.; Jones, J.R.; Trotter, J.H.; Chang, L.; O’Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical Manipulation of Hsp70 ATPase Activity Regulates Tau Stability. J. Neurosci. 2009, 29, 12079–12088, doi:10.1523/jneurosci.3345-09.2009.
- Yang, Y.; Turner, R.S.; Gaut, J.R. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 1998, 273, 25552–25555.
- Cappello, F.; Conway de Macario, E.; Marasa, L.; Zummo, G.; Macario, A.J. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809.
- Capello, F.; Gammazza, A.M.; Vilasi, S.; Ortore, M.G.; Biagio, P.L.S.; Campanella, C.; Pace, A.; Piccionello, A.P.; Taglialatela, G.; Macario, E.C.D.; et al. Chaperonotherapy for Alzheimer’s Disease: Focusing on HSP60. In Heat Shock Protein-Based Therapies; Asea, A.A.A., Almasoud, N.N., Krishnan, S., Kaur, P., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 51–77.
- Juwono, J.; Martinus, R.D. Does Hsp60 Provide a Link between Mitochondrial Stress and Inflammation in Diabetes Mellitus? J. Diabetes Res. 2016, 2016, 6, doi:10.1155/2016/8017571.
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258, doi:10.1006/bbrc.2000.4109.
- Veereshwarayya, V.; Kumar, P.; Rosen, K.M.; Mestril, R.; Querfurth, H.W. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J. Biol. Chem. 2006, 281, 29468–29478, doi:10.1074/jbc.M602533200.
- Xanthoudakis, S.; Roy, S.; Rasper, D.; Hennessey, T.; Aubin, Y.; Cassady, R.; Tawa, P.; Ruel, R.; Rosen, A.; Nicholson, D.W. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999, 18, 2049–2056, doi:10.1093/emboj/18.8.2049.
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 1999, 18, 2040–2048, doi:10.1093/emboj/18.8.2040.
- Walls, K.C.; Coskun, P.; Gallegos-Perez, J.L.; Zadourian, N.; Freude, K.; Rasool, S.; Blurton-Jones, M.; Green, K.N.; LaFerla, F.M. Swedish Alzheimer Mutation Induces Mitochondrial Dysfunction Mediated by HSP60 Mislocalization of Amyloid Precursor Protein (APP) and Beta-Amyloid. J. Biol. Chem. 2012, 287, 30317–30327, doi:10.1074/jbc.M112.365890.
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579, doi:10.1038/381571a0.
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2017, 69, 227-237.e224, doi:10.1016/j.molcel.2017.12.003.
- Alderson, T.R.; Kim, J.H.; Markley, J.L. Dynamical structures of Hsp70 and Hsp70–Hsp40 complexes. Structure 2016, 24, 1014–1030, doi:10.1016/j.str.2016.05.011.
- Abu-Farha, M.; Cherian, P.; Al-Khairi, I.; Tiss, A.; Khadir, A.; Kavalakatt, S.; Warsame, S.; Dehbi, M.; Behbehani, K.; Abubaker, J. DNAJB3/HSP-40 cochaperone improves insulin signaling and enhances glucose uptake in vitro through JNK repression. Sci. Rep. 2015, 5, 14448, doi:10.1038/srep14448.
- Haslbeck, M.; Franzmann, T.; Weinfurtner, D.; Buchner, J. Some like it hot: The structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005, 12, 842–846, doi:10.1038/nsmb993.
- Lee, G.J.; Roseman, A.M.; Saibil, H.R.; Vierling, E. A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 1997, 16, 659–671, doi:10.1093/emboj/16.3.659.
- Reddy, V.S.; Jakhotia, S.; Reddy, P.Y.; Reddy, G.B. Hyperglycemia induced expression, phosphorylation, and translocation of αB-crystallin in rat skeletal muscle. IUBMB Life 2015, 67, 291–299, doi:10.1002/iub.1370.
- Reddy, V.S.; Raghu, G.; Reddy, S.S.; Pasupulati, A.K.; Suryanarayana, P.; Reddy, G.B. Response of small heat shock proteins in diabetic rat retina. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7674–7682, doi:10.1167/iovs.13-12715.
- Shimura, H.; Miura-Shimura, Y.; Kosik, K.S. Binding of Tau to Heat Shock Protein 27 Leads to Decreased Concentration of Hyperphosphorylated Tau and Enhanced Cell Survival. J. Biol. Chem. 2004, 279, 17957–17962, doi:10.1074/jbc.M400351200.
- Mannini, B.; Cascella, R.; Zampagni, M.; van Waarde-Verhagen, M.; Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484, doi:10.1073/pnas.1117799109.
- Bakthisaran, R.; Tangirala, R.; Rao, C.M. Small heat shock proteins: Role in cellular functions and pathology. Biochim. Biophys. Acta BBA Proteins Proteom. 2015, 1854, 291–319, doi:https://doi.org/10.1016/j.bbapap.2014.12.019.
- Stege, G.J.; Renkawek, K.; Overkamp, P.S.; Verschuure, P.; van Rijk, A.F.; Reijnen-Aalbers, A.; Boelens, W.C.; Bosman, G.J.; de Jong, W.W. The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem. Biophys. Res. Commun. 1999, 262, 152–156, doi:10.1006/bbrc.1999.1167.
- Takino, J.-i.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655, doi:10.1007/s00535-009-0193-9.
- Hoshino, T.; Murao, N.; Namba, T.; Takehara, M.; Adachi, H.; Katsuno, M.; Sobue, G.; Matsushima, T.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s Disease-Related Phenotypes by Expression of Heat Shock Protein 70 in Mice. J. Neurosci. 2011, 31, 5225–5234, doi:10.1523/jneurosci.5478-10.2011.
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat Shock Protein 70 Prevents both Tau Aggregation and the Inhibitory Effects of Preexisting Tau Aggregates on Fast Axonal Transport. Biochemistry 2011, 50, 10300–10310, doi:10.1021/bi2009147.

