 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas R. Holmes | + 4078 word(s) | 4078 | 2020-11-13 05:19:24 | | | |
| 2 | Vivi Li | -70 word(s) | 4008 | 2020-11-25 07:17:26 | | |
Video Upload Options
Spherical nucleic acids (SNAs) are nanostructures consisting of nucleic acids in a spherical configuration, often around a nanoparticle core. SNAs are advantageous as gene-regulating agents compared to conventional gene therapy owing to their low toxicity, enhanced stability, uptake by virtually any cell, and ability to penetrate the epidermal barrier.
1. Introduction to SNAs: Structure and Synthesis
1.1. SNA Structure
1.1.1. SNA Core
Spherical nucleic acids are a class of gene modulatory molecules that share the spherical structure of densely oriented nucleic acids anchored to a central core. First described in 1996, the original SNAs were 60–80 thiol-conjugated DNA oligomers in a spherical orientation around a 13 nanometer (nm) gold nanoparticle core (originally called DNA Au-NPs) [1]. Since their initial creation with Au-NP cores, a wide variety of inorganic (silver, iron oxide, quantum dots, or silica) or organic materials (proteins, polymers, DNA, or liposomes) have been developed to serve as the SNA core [2][3][4][5][6]. Depending on the type of core used, magnetic, luminescent, electrical, catalytic, light scattering, and spectroscopic properties can be conferred upon SNAs for use as molecular probes or assembly into complex structures [7][8]. For example, magnetic and conductive properties of SNA cores allow them to be used in a wide range of microarray-based assays for detection of specific oligonucleotides or proteins [8][9][10][11][12][13][14]. Coreless SNAs have also been produced as self-assembling, spherically-oriented oligomers or, alternatively, by creating Au-NP SNAs, catalytic crosslinking of the functionalized oligo at the base of the strand closest to the core, and then dissolving the gold core using potassium cyanide, leaving behind a hollow SNA [15][16]. These coreless nanoparticles eliminate the potential long-term toxicity of the core (albeit low-risk) and highlight that the chemical and physical properties of SNAs, including their facile cellular uptake, are governed by the oligonucleotide conjugates themselves, rather than the core [17][18]. Au-NPs were originally chosen for the SNA core material because they are easy to synthesize, can be made highly monodisperse, can be conjugated to many types of ligands, have well-defined catalytic properties, and are readily tracked by inductively coupled mass spectrometry, all of which makes them a popular choice for SNA use in vitro.
1.1.2. Oligonucleotide Attachment to SNA Core
The oligonucleotides used for SNAs typically have three main components: (i) a terminal moiety that aids in its attachment to the core; (ii) a spacer region; and (iii) a specific functional region that is designed for target gene recognition. In the case of gold core SNAs, the oligonucleotide is commonly functionalized with a thiol group (termed alkylthiol-DNA/RNA) on one terminal end, facilitating the attachment of alkylthiol-DNA/RNA to the core surface via gold-sulfur bonds [1]. However, modification of the thiol moiety can enhance the oligo Au-NP interactions, including the use of cyclic disulfides or branched thiol groups [19][20]. When creating SNAs using silver nanoparticle cores (Ag-NPs), cyclic disulfides are preferable due to the more unstable surface of Ag-NPs [21]. Thiol-modified oligonucleotides are also used when making certain coreless, silica, or quantum dot core SNAs, in which Au-NPs are used as a scaffold [5][15][22]. Self-assembling or protein core SNA oligos contain a dibenzocyclooctone-amine (DBCO) moiety which facilitates its attachment to azide groups on polymers using click chemistry [16][23][24]. When generating liposomal SNAs, a tocopherol moiety is used for attachment to the liposomal core [5].
1.1.3. Oligonucleotide Spacer Region
Construction of the spacer region is also important to maximize SNA stability and oligonucleotide load. The spacer gives the conjugated oligonucleotide more flexibility and provides a buffer from the cationic core, which allows for more efficient hybridization with its target sequence. Spacers consisting of a 10 thymine/alanine sequence or polyethylene glycol (PEG) are commonly used due to their low interaction with the metal cores, thus increasing DNA/RNA load [25]. A polythymine spacer sequence has been shown to protect SNAs from salt-induced aggregation [26]. SNA oligos used for coreless SNAs contain modified nucleotides in the spacer region with an alkyne group, which allows for the catalytic crosslinking of nucleotide strands [15]. Another crosslinking method utilizes a thymine spacer with C6-amines which can be crosslinked with PEGylated bis(sulfosuccinimidyl)suberate [22].
1.1.4. Oligonucleotide Targeting Sequence
The conjugated oligonucleotide sequence that targets the gene of interest is located on the end farthest from where the oligonucleotide attaches to the nanoparticle core. Rosi et al. were the first to show that SNAs could enter a cell and knockdown gene expression using single-stranded DNA to target GFP expression [27]. Since then, duplexed DNA, siRNA or miRNA conjugates have been attached to SNA cores to regulate gene expression [18][28][29][30][31]. Catalytic RNAs, termed ribozymes, have been loaded onto SNAs to directly cleave the target mRNA strand as opposed to relying on the native RNA interference (RNAi) pathway for cleavage [32]. DNA can be loaded onto SNA cores at about 60–80 strands per core, whereas RNA has been loaded onto cores at about 30–45 RNA strands per core [29][30]. The inherent instability of RNA compared to DNA is a factor to consider when designing SNAs for gene regulation. To achieve similar stability to DNA-SNAs, RNA-SNAs can be passivated with PEG, which risks reducing cellular uptake, or the RNA is synthesized using modified nucleotides (i.e., 2′-O-methyl) to increase stability [33][34]. Phosphorothioate oligonucleotides, which replace the non-bridging oxygen in the natural phosphodiester bond of DNA/RNA backbones with a sulfur bond, increase oligomer stability and have been shown to promote their nuclear retention, allowing one to specifically target nuclear sequences, for example long non-coding RNAs [35]. Despite the challenges posed by RNA instability in SNA production, RNA-SNAs are still easily taken up by cells, have improved stability (when compared to linear RNA) through functionalization of RNA onto the solid core, can utilize the intracellular RNAi pathway machinery, and allow for more precise targeting, including with single base pair accuracy [28].
1.1.5. Oligonucleotide Functional Additives
Oligonucleotide conjugates can be functionalized with other groups to tailor their utilization. For example, fluorescent tags can be added to track cellular uptake or detect unique gene expression through attaching specialized “nanoflares”, dyes attached to detect cellular levels of analytes, or antibodies can be complexed with SNAs to detect proteins [6][12][25][36][37][38]. Whereas most SNAs aim to knock down expression of a specific gene of interest, nanoflares utilize a short, mRNA-targeting DNA sequence attached to a fluorophore that is quenched due to its close proximity to the nanoparticle surface to allow detection [39]. Upon binding to the mRNA target and dissolution from the nanoparticle, the fluorophore is no longer quenched and thus produces a signal [39]. A similar SNA technology, “sticky-flares”, uses longer antisense sequences that allow the fluorophore to remain bound to the transcript, enabling the tracking of mRNA movement in the cell and changes in gene expression in real time [40]. Rapidly proliferating cancer cells with high telomerase activity can be identified using SNA oligos functionalized with telomerase primers, while immune cells can be conditioned to target cancer cells by loading SNAs with cancer cell lysates or antigens [41][42][43]. Antibodies can be conjugated to the DNA or absorbed onto the SNA for cell-specific targeting [44]. SNAs have also been used to increase contrast in magnetic resonance imaging (MRI) to enhance uptake of Gd (III) chelates into cells [45]. Owing to their favorable properties of rapid cell uptake and stability, SNAs have also been used for drug delivery by loading chemotherapeutic agents onto the SNA shell or into the SNA core [6][46][47].
1.2. SNA Synthesis Using an Au-NP Core
Synthesis of SNAs is often initiated by first producing the core material. Au-NPs are a common core used for SNA production and in many cases act as a scaffold when synthesizing silica-core or coreless SNAs [5][15]. Au-NPs are typically synthesized by reducing the gold (III) chloride dihydrate through boiling in the presence of sodium citrate [48] (Figure 1a). This method of gold reduction, called the Frens method, produces gold particles in the range of 12–64 nm, depending on the molar ratio of citrate to gold [48]. The solution turns from yellow to gray to eventually a deep red, which indicates that the gold particles have been reduced [1]. There are several methods for Au-NP reduction that can produce particles of various sizes (reviewed in [49]). The smaller the diameter of the Au-NP core, the greater the density of oligonucleotide that can be loaded onto the Au-NP; however, the larger the nanoparticle, the more total oligomers that can be loaded (for example, one can load up to 600 strands on a 30 nm particle). This phenomenon is explained by the increased radial curvature of the smaller Au-NP, which increases the deflection angle between DNA, thus decreasing steric hindrance and electrostatic repulsion between strands [50]. The final concentration of Au-NPs is determined by measuring the absorbance (A) on a UV-visible spectrophotometer (for example, at a wavelength of ~520 nm for the gold nanoparticles) [25]. The concentration of Au-NP SNAs can then be determined using Beer’s Law (A = εLc) where A is the absorbance measured, ε is the extinction coefficient, which is based on the Au-NP diameter, and L is the path length (typically 1 cm) [22].
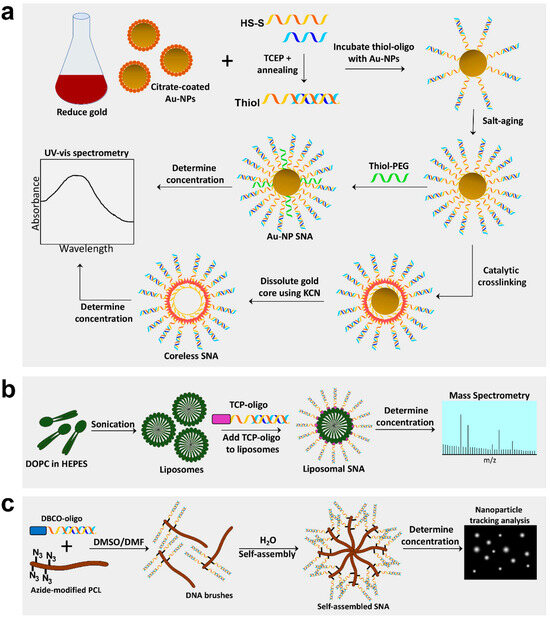
Figure 1. Synthesis of Au-NP core, coreless, liposomal core, and self-assembling spherical nucleic acids (SNAs). (a) Gold nanoparticle (Au-NP) SNAs are produced by reducing gold into 13 nm Au-NPs and preparing oligos by annealing sense and antisense strands and cleaving the disulfide bond to create a free thiol group. Au-NPs are incubated with thiol-oligos, salt-aged to increased oligo density, and thiol-polyethylene glycol (PEG) added to fill empty spaces. Lastly, Au-NP SNA concentration can be determined by spectrophotometry. Coreless SNAs are produced by crosslinking oligo strands on the surface of the Au-NP, then dissolving the gold core using potassium cyanide (KCN). (b) Liposomal SNAs are synthesized by sonicating 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) free fatty acids in HEPES buffer to create liposomes ~30 nm in diameter; liposomes are incubated with oligos containing a tocopherol (TCP) moiety for integration into liposomes. L-SNA concentration is determined by mass spectrometry. (c) Self-assembling SNAs are produced by conjugating oligos with a dibenzocyclooctone-amine (DBCO) moiety to polycaprolactone (PCL) polymers modified with azide groups in a DMSO/DMF solution to create DNA brush polymers. The DNA brushes then self-assemble in the presence of water and the concentration can be determined using nanoparticle tracking analysis.
After reduction, the citrate-coated Au-NPs are treated with diethyl pyrocarbonate and autoclaved to inhibit nucleotide degradation and sterilize the particles, respectively. This step is especially critical for the production of RNA-conjugated SNAs. Before adding the oligonucleotide to the Au-NPs, the oligo strands may need to be prepped for conjugation to the Au-NP core. For example, thiolated oligos (like those from Integrated DNA Technologies) are capped with a disulfide bond, so it must be reduced using tris(2-carboxyethyl)phosphine (TCEP) to expose a free thiol for attachment to the core. Additionally, if using duplexed thiol-DNA or RNA, a duplex formation reaction is performed to anneal the sense and antisense strands (single-stranded thiol-DNA or RNA skips this step). Once ready for Au-NP conjugation, single-stranded or hybridized thiol-oligonucleotide strands are added to the Au-NP solution at a ratio of ~20:1 (oligo:Au-NP) and incubated at room temp for about one hour (Figure 1a). Next, sodium salt is added to the DNA/RNA Au-NPs at a concentration of 50–150 mM, incubated overnight, and then slowly increased in 50 mM increments until the final concentration reaches 350 mM, a method called “salt-aging” [25][51] (Figure 1a). The negatively charged DNA repels the insertion of more DNA duplexes, so the addition of the positively charged sodium ions blocks these repulsions, allowing higher densities of DNA on the Au-NP core [51]. It is critical that salt-aging is performed gradually during a period of 1–2 days to prevent aggregation of SNAs. However, salt-aging can be hastened considerably by reducing the reaction pH to 3.0. Pre-treatment of Au-NPs with a surfactant (for example, Tween 20), reduces Au-NP aggregation during salt-aging and facilitates oligonucleotide attachment to the core [52]. The high DNA density and strong sulfur-gold interactions (which displaces the much weaker citrate-gold interactions) give Au-NP SNAs a negative zeta potential (<−30 mV), which ultimately helps prevent their aggregation [53]. After oligo conjugation to the Au-NP core, coreless SNAs can be produced by catalytic crosslinking of the oligos via the modified nucleotides in the spacer region, and then dissolving the gold core using potassium cyanide (Figure 1a).
Once the Au-NPs are fully saturated with oligonucleotides, any empty spaces remaining on the surface of the SNA core are backfilled with thiol-conjugated PEG to further improve stability. Bromide has also been used as a backfiller that can efficiently displace improperly attached oligos on the Au-NP surface [54]. The SNAs are concentrated using centrifugal filtration units and washed several times with 1× phosphate buffered saline to remove residual surfactants, salts, or unattached DNA/RNA which could inhibit downstream applications. The final concentration of Au-NP SNAs is determined by measuring the absorbance (A) on a UV-visible spectrophotometer and Beer’s Law (A = εLc) as described above. To determine the oligo concentration, the nucleotide strands can be dissociated from the gold core with dithiothreitol treatment, Au-NPs removed by centrifugation, and then absorbance measured on a UV-visible spectrophotometer at 260 nm. The concentrated SNAs can be used directly on cells in culture (both 2D and 3D) or topically in vivo, such as on mouse skin. As described above, SNA’s enhanced stability allows oligo SNAs to remain intact for months both at room and physiological temperatures.
The requirement for thiol conjugation of siRNAs to SNA cores for each targeting siRNA translates into highly expensive, specially ordered oligonucleotides. One approach to this problem is the use of universal SNAs that contain DNA anchors and a generated sense strand of siRNA that can be ligated to the DNA [55]. This also saves time, as new siRNA-SNAs can be produced through simple ligation and annealing reactions instead of full SNA production for each target.
1.3. Generation of Liposomal SNAs
Liposomal SNAs (L-SNA) are a newer SNA class using ~30 nm diameter unilamellar vesicles as the SNA core, prepared by sonication of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) in HEPES buffered saline [2] (Figure 1b). Oligonucleotides are attached to L-SNA cores using a tocopherol (TCP) moiety on the terminal end, which is absorbed by the liposomal core via hydrophobic interactions [2]. The TCP-oligos are incubated with liposomal cores at a ratio of 100:1 oligo to liposomes and the formed L-SNAs are then concentrated using tangential flow filtration with a 30 kD weight cutoff. Similarly to Au-NP SNAs, the oligo concentration can be determined by dissolving the liposomes in 90% methanol to dissociate the oligonucleotides from the core, and then absorbance is measured on a UV-visible spectrophotometer at 260 nm. The concentration of L-SNA liposomal cores can be determined using mass spectrometry (see supplementary methods in [5]).
Zhang et al. has created liposomal-based SNAs using biodegradable DNA-brush block copolymers (DBBC) [16]. Multiple oligonucleotides share a hydrophobic lipid tail, which can self-assemble into a micelle liposomal core. DBBC-based L-SNAs display increased oligonucleotide density (and thus a greater negative charge), higher melting temperature, and greater cellular uptake compared to linear DNA block copolymer L-SNAs [16]. DBBC-based L-SNAs are produced by modifying polymers with azide groups which allows for the attachment of DBCO-oligos in the presence of an organic solvent (i.e., DMSO, DMF) using click chemistry (Figure 1c) [16]. Replacing the organic solvent with water by dialysis initiates self-assembly of the DBBC polymers into a micelle-like SNA roughly 40 nm in diameter [16]. Unlike Au-NP SNAs, these self-assembled micelle SNAs cannot be quantified by spectrophotometry and thus must be measured using nanoparticle tracking analysis (Figure 1c) [56].
Modification of the oligonucleotide attachment moiety can impact the activity and distribution of L-SNAs. Attaching DNA sequences to L-SNAs using a low-affinity cholesterol tail (CHOL-LSNA) or high-affinity diacylglycerol lipid tail (DPPE-LSNA) reduces the inflammatory cytokine response in mice after intravenous injection compared to free DNA [57]. In vitro, CHOL-LSNAs are less readily taken up by cells compared to DPPE-LSNAs [58]. When CHOL- and DPPE-LSNAs were functionalized with the same immunostimulatory oligonucleotides, DPPE-LSNAs were more potent stimulators of macrophages [58]. Similar to coreless SNAs, use of L-SNAs avoids the possible long-term toxicity issues that accompany use of metal core SNAs. For this reason, L-SNAs are the preferred SNA subtype for preclinical and clinical studies.
2. Using SNAs for the Detection and Treatment of Skin Disease
Topical therapeutics for the treatment of skin disease are the gold standard for optimizing effective delivery to the skin and minimizing the risk of systemic side effects. Topical agents have largely been limited to small molecule inhibitors or stimulators, as nucleic acids cannot traditionally penetrate the epidermal barrier. However, SNAs can traverse the stratum corneum and stratum granulosum, sites of epidermal barrier function, and are detectable in the dermis [59], providing a unique opportunity to modify gene expression directly in skin towards more effective treatment and prevention of skin disease. In early studies as proof-of-principle, mouse skin was treated with EGFR-targeting Au-NP SNAs for 3 weeks, which resulted in decreased skin thickness and suppressed EGFR signaling [59]. SNAs can also be injected into skin lesions, given their easy accessibility.
2.1. Diabetic Wound Healing
Impaired wound healing is a common complication in patients with type 2 diabetes (T2D). Nearly 10% of all Americans are diagnosed with T2D or pre-T2D, and 25% of these individuals will develop a foot ulcer in their lifetime. However, few treatment options are available. The monosialic ganglioside GM3, located on the outer leaflet of the plasma membrane and the most prevalent sialylated glycosphingolipid in skin, and its synthesizing enzyme, GM3 synthase (GM3S), are critical regulators of insulin resistance in T2D [60][61][62]. Additionally, skin-specific GM3S knockout mice fed a high fat diet to induce a T2D state are resistant to impaired wound healing [60]. Thus, GM3S represents a prime target for the treatment of T2D impaired wound healing.
Using Au-NP siRNA-conjugated SNAs, Randeria et al. successfully targeted GM3S expression in 2D keratinocytes and diabetic mouse skin [31] (Table 1). In diabetic mice, GM3S-targeted SNAs significantly improved wound healing (as determined by epidermal gap closure), abetted granulation tissue formation, and increased insulin-like growth factor 1 receptor and EGFR signaling [31].
2.2. Nanoflares to Detect Abnormal Scar Formation
Most research using SNAs in skin focuses on epidermal and keratinocyte structure and function. However, one recent study investigated the use of specialized SNAs (nanoflares) to identify abnormal gene expression in cutaneous dermal fibroblasts seen in aberrant fibrotic scars [63]. As previously described, nanoflares are Au-NP SNAs functionalized with fluorescently labeled oligonucleotides that only produce a signal upon hybridization to their target mRNA [39].
Hypertrophic and keloid scars are the result of an abnormal fibrotic response to wound healing and are associated with excessive collagen production from increased expression of connective tissue growth factor (CTGF) and transforming growth factor β. Early identification of these scar types is critical for therapeutic intervention, and non-invasive techniques would be ideal for detection, given the role of trauma in causing these abnormal scars. Yeo and colleagues developed CTGF-recognizing nanoflares in hopes of identifying fibroblasts in wounds that could induce abnormal scar formation [63]. In 2D primary culture of normal, hypertrophic, and keloidal fibroblasts, CTGF nanoflares accurately labelled disease fibroblasts that were overexpressing CTGF [63]. Live in vivo imaging detected significantly more nanoflares in mice injected with hypertrophic fibroblasts (HSFs) and CTGF nanoflares compared to those injected with normal fibroblasts (NDFs) and CTGF nanoflares [63]. Topical application of CTGF nanoflares was also successful at identifying hypertrophic fibroblasts in ex vivo human skin injected with NDFs or HSFs. Finally, CTGF nanoflares were able to distinguish scarred rabbit ears (post-wounding) from normal, unwounded rabbit ear skin after 48 h of topical application [63]. This research highlights how SNAs can be adapted for both gene regulation and detecting gene expression. Given the ability of SNAs to penetrate skin, this technology could be further adapted for noninvasive detection after topical application to human skin.
2.3. Psoriasis
Psoriasis is a common inflammatory skin disorder that is known to result from skewed immune activation towards the Th17/interleukin (IL)-23 and TNFα signaling pathways. While the treatment for patients with moderate-to-severe disease has been revolutionized in both efficacy and safety by the commercial availability of monoclonal antibodies targeting these pathways, the inability of these large monoclonal antibodies to traverse the epidermal barrier has prevented transition of this targeted approach to the 80% of patients with mild-to-moderate psoriasis.
Indeed, using liposomal SNAs (L-SNAs) with DNA targeting TNFA (AST-005, generated by Exicure, Inc., Chicago, IL 60614, USA), Lewandowski et al. showed uptake of Cy-5 fluorescent L-SNAs into the markedly thickened human psoriatic skin explants [64]. Using a 3D human raft model of psoriasis, generated by the addition of a cytokine cocktail (TNFα, IL-17 and IL-22), addition of AST-005 to the surface of the raft (simulating topical delivery) led to improvement in the differentiation abnormality of the psoriatic rafts and normalization in the mRNA expression of a variety of psoriatic markers (increases in TNFA, DEFB4, and S100A7, with decreases in KRT10 and LOR, all p < 0.001). TNFA LSNAs, applied topically every other day in a common moisturizer without penetration enhancers, reversed the development of psoriasis-like disease in the widely used imiquimod-induced mouse model [64]. After just a week of treatment mice showed improvement vs. vehicle and scrambled controls in modified Psoriasis Area and Severity Index scores (epidermal thickness and proliferation, immune cell infiltration, and psoriatic expression markers (all p < 0.001)) that was indistinguishable from mice without the psoriatic disease [64].
More recently, an L-SNA targeting IL17RA was tested in normal human skin explants, as well as in human 3D and imiquimod-induced mouse models of psoriasis [65]. The L-SNA knocked down IL17RA protein by 66% and 72% in the human normal explant and psoriatic 3D raft models, respectively. In these rafts, expression of psoriatic markers of Th17 pathway activation were also significantly reduced (IL17C by 85%, DEFB4 by 84%, TNFA by 77%, and PI3 by 65%) in comparison with scrambled and vehicle controls (all p < 0.001). An L-SNA targeting IL17RA in the imiquimod-induced mouse model reduced the modified Psoriasis Area and Severity Index by 74% vs. vehicle-treated mice, reduced epidermal thickness by 56%, and, as in the 3D models, decreased mRNA expression of the various psoriasis markers [65].
These results demonstrated the efficacy of SNAs in penetrating even thickened skin, knocking down gene targets, and reversing skin disease in mouse and human 3D culture models of psoriasis. Studies are ongoing with bispecific L-SNAs that target both TNFα and IL17RA.
3. Conclusions
The impact and potential use of SNAs in both research and clinical applications has helped transform the mechanisms of gene regulation. However, some challenges do exist. Serum proteins are attracted to the oligonucleotide shell of SNAs forming a protein corona [66]. This is especially true of glycine-rich sequences, in which the corona has been shown to increase the rate of macrophage-mediated SNA removal and accumulation in the liver and spleen in mice, thus preventing their uptake by target cells [66][67]. Coating SNAs with PEG to prevent protein corona formation on SNAs has proven beneficial, but it can also reduce cellular uptake [33]. More work will be needed to engineer SNAs to both elude macrophage clearance and maintain a high level of cellular uptake. Once inside the cell, the mechanisms of SNA endosomal/lysosomal release, and ways to maximize this release, will require more attention. Chemical additions on SNAs to improve intracellular trafficking have been developed, but these added chemicals increase the risk of toxicity. Despite progress in achieving SNA cell specificity, the ability of SNAs to be taken up by many cell types handicaps their ability to be cell-type specific with systemic delivery without off-target effects, emphasizing their value when topically applied or injected for skin disease. Lastly, although SNAs have little to no short-term toxicity, the potential long-term toxicity of SNAs, specifically with metal cores, is poorly understood. Coreless or more natural (i.e., liposomal) SNAs may address this concern, but more work is needed.
Despite these challenges, SNAs have made substantial progress both in basic research and clinical use. This is especially true in the field of dermatology because skin offers a unique opportunity to side-step some of the challenges SNAs pose. For example, skin pathologies, being that they are on the skin’s surface, are more easily accessible to drugs. For this reason, using SNAs directly on the skin largely removes the possibility of systemic toxicity or off-target effects. Along the same lines, SNAs topically applied to the skin avoid using the bloodstream, where they are more prone to immune clearance or degradation, to reach their target. Knowing this, combined with the fact that SNAs can penetrate the skin without the use of secondary reagents, SNA use for gene modulation in dermatology is a seemingly perfect match. Continued development of SNAs can expand the use of gene modulation techniques for the topical intervention of cutaneous disease.
References
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609.
- Banga, R.J.; Chernyak, N.; Narayan, S.P.; Nguyen, S.T.; Mirkin, C.A. Liposomal spherical nucleic acids. J. Am. Chem. Soc. 2014, 136, 9866–9869.
- Brodin, J.D.; Auyeung, E.; Mirkin, C.A. DNA-mediated engineering of multicomponent enzyme crystals. Proc. Natl. Acad. Sci. USA 2015, 112, 4564–4569.
- Ruan, W.; Zheng, M.; An, Y.; Liu, Y.; Lovejoy, D.B.; Hao, M.; Zou, Y.; Lee, A.; Yang, S.; Lu, Y.; et al. DNA nanoclew templated spherical nucleic acids for siRNA delivery. Chem. Commun. 2018, 54, 3609–3612.
- Young, K.L.; Scott, A.W.; Hao, L.; Mirkin, S.E.; Liu, G.; Mirkin, C.A. Hollow spherical nucleic acids for intracellular gene regulation based upon biocompatible silica shells. Nano Lett. 2012, 12, 3867–3871.
- Zhu, S.; Xing, H.; Gordiichuk, P.; Park, J.; Mirkin, C.A. PLGA spherical nucleic acids. Adv. Mater. 2018, 30, e1707113.
- Park, S.S.; Urbach, Z.J.; Brisbois, C.A.; Parker, K.A.; Partridge, B.E.; Oh, T.; Dravid, V.P.; Olvera de la Cruz, M.; Mirkin, C.A. DNA- and field-mediated assembly of magnetic nanoparticles into high-aspect ratio crystals. Adv. Mater. 2020, 32, e1906626.
- Taton, T.A.; Mirkin, C.A.; Letsinger, R.L. Scanometric DNA array detection with nanoparticle probes. Science 2000, 289, 1757–1760.
- Cao, Y.C.; Jin, R.; Nam, J.M.; Thaxton, C.S.; Mirkin, C.A. Raman dye-labeled nanoparticle probes for proteins. J. Am. Chem. Soc. 2003, 125, 14676–14677.
- Hong, M.; Sun, H.; Xu, L.; Yue, Q.; Shen, G.; Li, M.; Tang, B.; Li, C.Z. In situ monitoring of cytoplasmic precursor and mature microRNA using gold nanoparticle and graphene oxide composite probes. Anal. Chim. Acta 2018, 1021, 129–139.
- Hong, M.; Zhou, X.; Lu, Z.; Zhu, J. Nanoparticle-based, fluorous-tag-driven DNA detection. Angew. Chem. Int. Ed. Engl. 2009, 48, 9503–9506.
- Nam, J.M.; Thaxton, C.S.; Mirkin, C.A. Nanoparticle-based bio-bar codes for the ultrasensitive detection of proteins. Science 2003, 301, 1884–1886.
- Park, S.J.; Lazarides, A.A.; Mirkin, C.A.; Brazis, P.W.; Kannewurf, C.R.; Letsinger, R.L. The electrical properties of gold nanoparticle assemblies linked by DNA. Angew. Chem. Int. Ed. Engl. 2000, 39, 3845–3848.
- Park, S.J.; Taton, T.A.; Mirkin, C.A. Array-based electrical detection of DNA with nanoparticle probes. Science 2002, 295, 1503–1506.
- Cutler, J.I.; Zhang, K.; Zheng, D.; Auyeung, E.; Prigodich, A.E.; Mirkin, C.A. Polyvalent nucleic acid nanostructures. J. Am. Chem. Soc. 2011, 133, 9254–9257.
- Zhang, C.; Hao, L.; Calabrese, C.M.; Zhou, Y.; Choi, C.H.; Xing, H.; Mirkin, C.A. Biodegradable DNA-brush block copolymer spherical nucleic acids enable transfection agent-free intracellular gene regulation. Small 2015, 11, 5360–5368.
- Barnaby, S.N.; Perelman, G.A.; Kohlstedt, K.L.; Chinen, A.B.; Schatz, G.C.; Mirkin, C.A. Design considerations for rna spherical nucleic acids (SNAs). Bioconjugate Chem. 2016, 27, 2124–2131.
- Melamed, J.R.; Kreuzberger, N.L.; Goyal, R.; Day, E.S. Spherical nucleic acid architecture can improve the efficacy of polycation-mediated siRNA delivery. Mol. Ther. Nucleic Acids 2018, 12, 207–219.
- Li, Z.; Jin, R.; Mirkin, C.A.; Letsinger, R.L. Multiple thiol-anchor capped DNA-gold nanoparticle conjugates. Nucleic Acids Res. 2002, 30, 1558–1562.
- Dougan, J.A.; Karlsson, C.; Smith, W.E.; Graham, D. Enhanced oligonucleotide-nanoparticle conjugate stability using thioctic acid modified oligonucleotides. Nucleic Acids Res. 2007, 35, 3668–3675.
- Cao, Y.; Jin, R.; Mirkin, C.A. DNA-modified core-shell Ag/Au nanoparticles. J. Am. Chem. Soc. 2001, 123, 7961–7962.
- Banga, R.J.; Meckes, B.; Narayan, S.P.; Sprangers, A.J.; Nguyen, S.T.; Mirkin, C.A. Cross-Linked Micellar Spherical Nucleic Acids from Thermoresponsive Templates. J. Am. Chem. Soc. 2017, 139, 4278–4281.
- Kusmierz, C.D.; Bujold, K.E.; Callmann, C.E.; Mirkin, C.A. Defining the Design Parameters for in Vivo Enzyme Delivery Through Protein Spherical Nucleic Acids. ACS Cent. Sci. 2020, 6, 815–822.
- Li, H.; Zhang, B.; Lu, X.; Tan, X.; Jia, F.; Xiao, Y.; Cheng, Z.; Li, Y.; Silva, D.O.; Schrekker, H.S.; et al. Molecular spherical nucleic acids. Proc. Natl. Acad. Sci. USA 2018, 115, 4340–4344.
- Hurst, S.J.; Lytton-Jean, A.K.; Mirkin, C.A. Maximizing DNA loading on a range of gold nanoparticle sizes. Anal. Chem. 2006, 78, 8313–8318.
- Sun, J.; Curry, D.; Yuan, Q.; Zhang, X.; Liang, H. Highly hybridizable spherical nucleic acids by tandem glutathione treatment and polythymine spacing. ACS Appl. Mater. Interfaces 2016, 8, 12504–12513.
- Rosi, N.L.; Giljohann, D.A.; Thaxton, C.S.; Lytton-Jean, A.K.; Han, M.S.; Mirkin, C.A. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science 2006, 312, 1027–1030.
- Giljohann, D.A.; Seferos, D.S.; Prigodich, A.E.; Patel, P.C.; Mirkin, C.A. Gene regulation with polyvalent siRNA-nanoparticle conjugates. J. Am. Chem. Soc. 2009, 131, 2072–2073.
- Hao, L.; Patel, P.C.; Alhasan, A.H.; Giljohann, D.A.; Mirkin, C.A. Nucleic acid-gold nanoparticle conjugates as mimics of microRNA. Small 2011, 7, 3158–3162.
- Oishi, M.; Nakaogami, J.; Ishii, T.; Nagasaki, Y. Smart PEGylated gold nanoparticles for the cytoplasmic delivery of siRNA to induce enhanced gene silencing. Chem. Lett. 2006, 35, 1046–1047.
- Randeria, P.S.; Seeger, M.A.; Wang, X.Q.; Wilson, H.; Shipp, D.; Mirkin, C.A.; Paller, A.S. siRNA-based spherical nucleic acids reverse impaired wound healing in diabetic mice by ganglioside GM3 synthase knockdown. Proc. Natl. Acad. Sci. USA 2015, 112, 5573–5578.
- Rouge, J.L.; Sita, T.L.; Hao, L.; Kouri, F.M.; Briley, W.E.; Stegh, A.H.; Mirkin, C.A. Ribozyme-spherical nucleic acids. J. Am. Chem. Soc. 2015, 137, 10528–10531.
- Chinen, A.B.; Ferrer, J.R.; Merkel, T.J.; Mirkin, C.A. Relationships between poly(ethylene glycol) modifications on RNA-spherical nucleic acid conjugates and cellular uptake and circulation time. Bioconjugate Chem. 2016, 27, 2715–2721.
- Barnaby, S.N.; Lee, A.; Mirkin, C.A. Probing the inherent stability of siRNA immobilized on nanoparticle constructs. Proc. Natl. Acad. Sci. USA 2014, 111, 9739–9744.
- Sprangers, A.J.; Hao, L.; Banga, R.J.; Mirkin, C.A. Liposomal spherical nucleic acids for regulating long noncoding rnas in the nucleus. Small 2017, 13, e1602753.
- Song, Q.; Wang, X.Q.; Holmes, T.R.; Briley, W.; Roth, E.W.; Ponedal, A.; Bonkowski, M.; Mirkin, C.; Paller, A.S. Epidermal class A scavenger receptor complexes are lipid raft-based and promote nucleic acid nanoparticle uptake. J. Investig. Dermatol. 2020, in press.
- Kim, D.; Daniel, W.L.; Mirkin, C.A. Microarray-based multiplexed scanometric immunoassay for protein cancer markers using gold nanoparticle probes. Anal. Chem. 2009, 81, 9183–9187.
- Samanta, D.; Ebrahimi, S.B.; Kusmierz, C.D.; Cheng, H.F.; Mirkin, C.A. Protein Spherical Nucleic Acids for Live-Cell Chemical Analysis. J. Am. Chem. Soc. 2020, 142, 13350–13355.
- Seferos, D.S.; Giljohann, D.A.; Hill, H.D.; Prigodich, A.E.; Mirkin, C.A. Nano-flares: Probes for transfection and mRNA detection in living cells. J. Am. Chem. Soc. 2007, 129, 15477–15479.
- Briley, W.E.; Bondy, M.H.; Randeria, P.S.; Dupper, T.J.; Mirkin, C.A. Quantification and real-time tracking of RNA in live cells using Sticky-flares. Proc. Natl. Acad. Sci. USA 2015, 112, 9591–9595.
- Callmann, C.E.; Cole, L.E.; Kusmierz, C.D.; Huang, Z.; Horiuchi, D.; Mirkin, C.A. Tumor cell lysate-loaded immunostimulatory spherical nucleic acids as therapeutics for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 17543–17550.
- Liu, Z.; Zhao, J.; Zhang, R.; Han, G.; Zhang, C.; Liu, B.; Zhang, Z.; Han, M.Y.; Gao, X. Cross-Platform Cancer Cell Identification Using Telomerase-Specific Spherical Nucleic Acids. ACS Nano 2018, 12, 3629–3637.
- Qin, L.; Wang, S.; Dominguez, D.; Long, A.; Chen, S.; Fan, J.; Ahn, J.; Skakuj, K.; Huang, Z.; Lee, A.; et al. Development of Spherical Nucleic Acids for Prostate Cancer Immunotherapy. Front. Immunol. 2020, 11, 1333.
- Zhang, W.; Meckes, B.; Mirkin, C.A. Spherical nucleic acids with tailored and active protein coronae. ACS Cent. Sci. 2019, 5, 1983–1990.
- Song, Y.; Xu, X.; MacRenaris, K.W.; Zhang, X.Q.; Mirkin, C.A.; Meade, T.J. Multimodal gadolinium-enriched DNA-gold nanoparticle conjugates for cellular imaging. Angew. Chem. Int. Ed. Engl. 2009, 48, 9143–9147.
- Bousmail, D.; Amrein, L.; Fakhoury, J.J.; Fakih, H.H.; Hsu, J.C.C.; Panasci, L.; Sleiman, H.F. Precision spherical nucleic acids for delivery of anticancer drugs. Chem. Sci. 2017, 8, 6218–6229.
- Wang, S.; Qin, L.; Yamankurt, G.; Skakuj, K.; Huang, Z.; Chen, P.C.; Dominguez, D.; Lee, A.; Zhang, B.; Mirkin, C.A. Rational vaccinology with spherical nucleic acids. Proc. Natl. Acad. Sci. USA 2019, 116, 10473–10481.
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature 1973, 241, 20–22.
- Hayat, M.A. Colloidal Gold: Principles, Methods, and Applications; Academic Press: Cambridge, MA, USA, 1989; pp. 13–32.
- Hill, H.D.; Millstone, J.E.; Banholzer, M.J.; Mirkin, C.A. The role radius of curvature plays in thiolated oligonucleotide loading on gold nanoparticles. ACS Nano 2009, 3, 418–424.
- Elghanian, R.; Storhoff, J.J.; Mucic, R.C.; Letsinger, R.L.; Mirkin, C.A. Selective colorimetric detection of polynucleotides based on the distance-dependent optical properties of gold nanoparticles. Science 1997, 277, 1078–1081.
- Zhao, Y.; Wang, Z.; Zhang, W.; Jiang, X. Adsorbed Tween 80 is unique in its ability to improve the stability of gold nanoparticles in solutions of biomolecules. Nanoscale 2010, 2, 2114–2119.
- Giljohann, D.A.; Seferos, D.S.; Patel, P.C.; Millstone, J.E.; Rosi, N.L.; Mirkin, C.A. Oligonucleotide loading determines cellular uptake of DNA-modified gold nanoparticles. Nano Lett. 2007, 7, 3818–3821.
- Liu, B.; Wu, P.; Huang, Z.; Ma, L.; Liu, J. Bromide as a Robust Backfiller on Gold for Precise Control of DNA Conformation and High Stability of Spherical Nucleic Acids. J. Am. Chem. Soc. 2018, 140, 4499–4502.
- Rouge, J.L.; Hao, L.; Wu, X.A.; Briley, W.E.; Mirkin, C.A. Spherical nucleic acids as a divergent platform for synthesizing RNA-nanoparticle conjugates through enzymatic ligation. ACS Nano 2014, 8, 8837–8843.
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm Res 2010, 27, 796–810.
- Ferrer, J.R.; Sinegra, A.J.; Ivancic, D.; Yeap, X.Y.; Qiu, L.; Wang, J.J.; Zhang, Z.J.; Wertheim, J.A.; Mirkin, C.A. Structure-dependent biodistribution of liposomal spherical nucleic acids. ACS Nano 2020, 14, 1682–1693.
- Meckes, B.; Banga, R.J.; Nguyen, S.T.; Mirkin, C.A. Enhancing the stability and immunomodulatory activity of liposomal spherical nucleic acids through lipid-tail DNA modifications. Small 2018, 14, e1702909.
- Zheng, D.; Giljohann, D.A.; Chen, D.L.; Massich, M.D.; Wang, X.Q.; Iordanov, H.; Mirkin, C.A.; Paller, A.S. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 11975–11980.
- Wang, X.Q.; Lee, S.; Wilson, H.; Seeger, M.; Iordanov, H.; Gatla, N.; Whittington, A.; Bach, D.; Lu, J.Y.; Paller, A.S. Ganglioside GM3 depletion reverses impaired wound healing in diabetic mice by activating IGF-1 and insulin receptors. J. Investig. Dermatol. 2014, 134, 1446–1455.
- Tagami, S.; Inokuchi Ji, J.; Kabayama, K.; Yoshimura, H.; Kitamura, F.; Uemura, S.; Ogawa, C.; Ishii, A.; Saito, M.; Ohtsuka, Y.; et al. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J. Biol. Chem. 2002, 277, 3085–3092.
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, J.L.; Werth, N.; et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449.
- Yeo, D.C.; Wiraja, C.; Paller, A.S.; Mirkin, C.A.; Xu, C. Abnormal scar identification with spherical-nucleic-acid technology. Nat. Biomed. Eng. 2018, 2, 227–238.
- Lewandowski, K.T.; Thiede, R.; Guido, N.; Daniel, W.L.; Kang, R.; Guerrero-Zayas, M.I.; Seeger, M.A.; Wang, X.Q.; Giljohann, D.A.; Paller, A.S. Topically delivered tumor necrosis factor-alpha-targeted gene regulation for psoriasis. J. Investig. Dermatol. 2017, 137, 2027–2030.
- Liu, H.; Kang, R.S.; Bagnowski, K.; Yu, J.M.; Radecki, S.; Daniel, W.L.; Anderson, B.R.; Nallagatla, S.; Schook, A.; Agarwal, R.; et al. Targeting the IL-17 receptor using liposomal spherical nucleic acids as topical therapy for psoriasis. J. Investig. Dermatol. 2020, 140, 435–444.
- Chinen, A.B.; Guan, C.M.; Mirkin, C.A. Spherical nucleic acid nanoparticle conjugates enhance G-quadruplex formation and increase serum protein interactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 527–531.
- Chinen, A.B.; Guan, C.M.; Ko, C.H.; Mirkin, C.A. The impact of protein corona formation on the macrophage cellular uptake and biodistribution of spherical nucleic acids. Small 2017, 13, e1603847.



