Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lăcrămioara Lacramioara Ionela Butnariu | -- | 3640 | 2022-10-29 07:31:57 | | | |
| 2 | Camila Xu | Meta information modification | 3640 | 2022-10-31 09:39:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Butnariu, L.I.; Gorduza, E.V.; Florea, L.; Țarcă, E.; Moisă, �.M.; Trandafir, L.M.; Stoleriu, S.; Bădescu, M.C.; Luca, A.; Popa, S.; et al. RAS/RAF/MAPK/ERK Signaling Pathways of Vascular Anomalies. Encyclopedia. Available online: https://encyclopedia.pub/entry/31857 (accessed on 04 August 2026).
Butnariu LI, Gorduza EV, Florea L, Țarcă E, Moisă �M, Trandafir LM, et al. RAS/RAF/MAPK/ERK Signaling Pathways of Vascular Anomalies. Encyclopedia. Available at: https://encyclopedia.pub/entry/31857. Accessed August 04, 2026.
Butnariu, Lăcrămioara Ionela, Eusebiu Vlad Gorduza, Laura Florea, Elena Țarcă, Ștefana Maria Moisă, Laura Mihaela Trandafir, Simona Stoleriu, Minerva Codruța Bădescu, Alina-Costina Luca, Setalia Popa, et al. "RAS/RAF/MAPK/ERK Signaling Pathways of Vascular Anomalies" Encyclopedia, https://encyclopedia.pub/entry/31857 (accessed August 04, 2026).
Butnariu, L.I., Gorduza, E.V., Florea, L., Țarcă, E., Moisă, �.M., Trandafir, L.M., Stoleriu, S., Bădescu, M.C., Luca, A., Popa, S., Radu, I., & Cojocaru, E. (2022, October 29). RAS/RAF/MAPK/ERK Signaling Pathways of Vascular Anomalies. In Encyclopedia. https://encyclopedia.pub/entry/31857
Butnariu, Lăcrămioara Ionela, et al. "RAS/RAF/MAPK/ERK Signaling Pathways of Vascular Anomalies." Encyclopedia. Web. 29 October, 2022.
Copy Citation
Vascular anomalies (VAs) are morphogenesis defects of the vascular system (arteries, capillaries, veins, lymphatic vessels) singularly or in complex combinations, sometimes with a severe impact on the quality of life. The main function of the RAS/RAF/MAPK/ERK signaling pathway, also called the “proliferation pathway”, is to transduce signals from the extracellular milieu to the cell nucleus where specific genes are activated for cell cycle regulation, proliferation, and cell migration.
vascular anomalies
signaling pathway
somatic mutation
1. Introduction
Vascular anomalies (VAs) represent a heterogeneous group of blood vessel anomalies: capillaries, lymphatics, arteries, veins, or a spectrum of mixed anomalies, the severity of which varies from a simple birthmark to severe, life-threatening entities [1]. The estimated prevalence of VAs is 4.5%, and most appear sporadically and isolated, but there are also syndromic forms of the disease [1]. In 1982, Mulliken and Glowacki described the first clinical classification of VAs that also considered the histological characteristics of vascular endothelial cells (ECs). This classification, later adopted by the International Society for the Study of Vascular Anomalies in 1996, classifies and differentiates VAs into two types: vascular proliferative tumors and vascular malformations [2][3]. The distinction between the two entities is based on histopathological assessment of increased ECs turnover. Vascular tumors (most of which are hemangiomas) are histologically characterized by a high turnover of ECs. They represent the neoplastic growth of vascular ECs and usually have an evolution that includes a proliferative phase, a period of plateau or stability, followed by spontaneous regression. Vascular malformations are anomalies of dysmorphogenesis composed of vascular channels with an abnormal structure, lined by ECs, without their proliferation and a normal turnover of vascular ECs. They are congenital, but sometimes they are not noticed immediately at birth. They do not regress, and grow proportionally with the development of the individual (Table 1) [2].
Table 1. Classification of Vascular Anomalies According to ISSVA (2018) [2].
| Vascular Anomalies | |
|---|---|
| Vascular Tumors | Benign |
| Locally aggressive or Borderline | |
| Malignant | |
| Vascular Malformations | |
| Simple | CM/VM/LM/AVM */AVF * |
| Combined | Defined as ≥2 vascular malformations found in one lesion |
| Anomalies of major named vessels (“channel type” or “truncal” vascular malformations) | Abnormalities in the origin/course/number/lenght/diameter (aplasia, hypoplasia, stenosis, ectasia/aneurysm)/persistence (of embrional vessels)/communication (AVF) of major blood vessels that have anatomical names |
| Associated with other anomalies | Syndromes that associate vascular malformations with non-vascular symptoms |
ISSVA: International Society for the Study of Vascular Anomalies; CM: Capillary malformation VM: Venous malformation; LM: Lymphatic malformation; AVM: Arteriovenous malformation; AVF: Arteriovenous fistula; * high-flow lesions.
Most of the VAs are present at birth or in the neonatal period and can develop later, but they can also appear later during the life of affected individuals. Etiologically, VAs are caused by inherited germline mutations or somatic mutations [3][4][5]. According to Knudson’s two-hit hypothesis, the second loss-of-function (LOF) mutation in somatic cells (in which a first mutation, inherited or acquired, is already present) will cause the loss of heterozygosity, the consequence being the LOF of the encoded protein in the affected cells and the appearance of VAs [3]. Most isolated VAs are caused by somatic gain-of-function (GOF) mutations of genes involved in angiogenesis, lymphangiogenesis, vascular cell proliferation and apoptosis, some of which are also detected in certain types of cancer [4].
The extreme phenotypic variability of VAs is correlated with the complex molecular mechanisms that involve numerous genes encoding molecules that intervene in different signaling pathways at the level of vascular cells: RAS (rat sarcoma)/RAF (rapidly accelerated fibrosarcoma)/MAPK (mitogen-activated protein kinase kinase)/ERK (extracellular signal-regulated kinase); angiopoietin/TIE2 (angiopoietin-1 receptor), PI3K (phosphoinositide 3-kinase)/AKT (protein kinase B)/mTOR (mammalian target of Rapamycin); TGFB (transforming growth factor beta) signaling, and the G protein–coupled receptor signaling molecules (GNA [G protein subunit alpha] Q/GNA11/GNA14) Figure 1) [4][5].
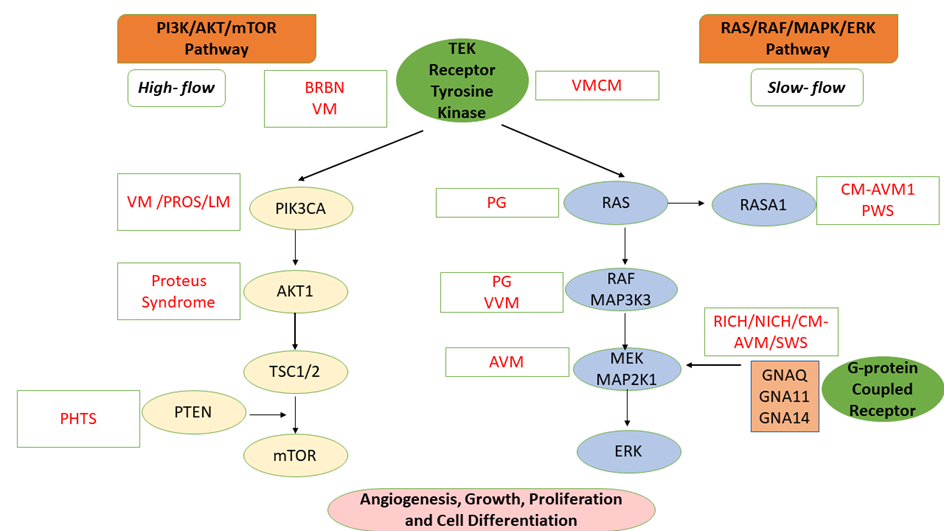
Figure 1. Molecular Mechanism of Vascular Anomalies: PI3K/AKT/mTOR and RAS/MAPK/ERK Signaling Pathways [1][4][5]. VM: Venous malformation; LM: Lymphatic malformation; AVM: Arteriovenous malformation; BRBN: Blue rubber bleb nevus (BRBN) syndrome; PHTS: PTEN hamartoma tumor syndrome; CM-AVM: Capillary malformation—Arteriovenous malformation syndrome; PROS: PIK3CA-related overgrowth spectrum; RICH: Rapidly involuting congenital hemangioma; NICH: Noninvoluting congenital hemangioma; VMCM: Cutaneomucosal venous malformation; PWS: Parkes Weber syndrome; VVM: Verrucous venous malformation; PG: Pyogenic granuloma; SWS: Sturge-Weber syndrome; MAP2K1: Mitogen-activated protein kinase kinase 1; ERK: Extracellular signal-regulated kinase; mTOR: Mammalian target of Rapamycin; PTEN: Phosphatase and tensin homolog; GNAQ: Guanine Nucleotide-Binding Protein G(Q) Subunit Alpha; TSC1/2: Tuberous sclerosis complex 1/2; TEK: TEK tyrosine kinase; RAF: Rapidly accelerated fibrosarcoma; PIK3CA: Phosphatidylinositol 4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha; RASA1: RAS p21 protein activator 1.
2. Vascular Anomalies: RAS/RAF/MAPK/ERK Signaling Pathways (RASopathies)
The main function of the RAS/RAF/MAPK/ERK signaling pathway, also called the “proliferation pathway”, is to transduce signals from the extracellular milieu to the cell nucleus where specific genes are activated for cell cycle regulation, proliferation, and cell migration. The mutations of these genes determine a group of diseases, called RASopathies, which associate with VAs [4][6].
Numerous growth factors, cytokines, and G protein–coupled receptor ligands induce signaling in the RAS/RAF/MAPK/ERK pathway, activating RAS by replacing GDP (Guanosine diphosphate) with GTP (Guanosine-5’-triphosphate). After binding to RAS, RAF exhibits serine/threonine-protein kinase activity and activates MAPK, which will subsequently activate ERK by phosphorylation [4].
The RAS/RAF/MAPK/ERK signaling pathway is also involved in cell cycle regulation, cell damage repair, integrin signaling, and can stimulate angiogenesis by altering the expression of genes directly involved in the formation of new blood vessels. Gene mutations involved in the RAS/RAF/MAPK/ERK pathway are correlated with tumorigenesis, the RAS gene being an oncogene whose mutations are frequently detected in human cancers [6][7].
2.1. Venous Malformations (VMs) and RAS/RAF/MAPK/ERK Signaling Pathway
Venous malformations (VMs) are slow-flow vascular lesions, caused by a defect in vascular morphogenesis during early embryonic life (weeks 4–10 of gestation), characterized by the cluster of dilated venous channels associated with a thin or absent vascular wall [8].
2.1.1. Verrucous Venous Malformation (VVM)
Verrucous Venous Malformation (VVM) also called Formerly Verrucous Hemangioma, is a non-hereditary venous malformation, caused by activating somatic mutations (ASM) of the MAP3K3 (Mitogen-Activated Protein Kinase Kinase Kinase 3) gene (OMIM 602539), located on chromosome 17q23.3 [9]. MAP3K3 is involved in both the ERK and the AKT/mTOR signaling pathways. VVMs are clinically manifested by cutaneous capillary venous malformations (CVMs) present as well-demarcated purpuric linear plaques covered by a hyperkeratotic dermis, which can reach sizes of several centimeters. VVMs can be present at birth, or appear early in childhood, most frequently being located at the level of the lower limbs. In young patients, the lesions have a red-blue appearance, are soft, but become hyperkeratotic over time [8][10].
2.1.2. Cavernous Cerebral Malformation (CCM)
Cerebral cavernous venous malformations (CCMs), also known as cavernous hemangiomas or cavernomas, are slow-flow VMs consisting of a “mulberry-like” cluster of hyalinized dilated thin-walled capillaries, without intervening normal brain tissue. CCMs affects up to 0.5% of the general population [8]. Due to recurrent microhemorrhages and thrombosis, they are usually surrounded by hemosiderin deposits and gliosis. The supratentorial location is most common, although lesions may also occur in the basal ganglia, brainstem, cerebellum, and spinal column. Occasionally, CCMs may be associated with a developmental venous anomaly (VAV), in which case they are known as a mixed vascular malformation [11].
Most cases appear sporadically and are represented by single lesions, which become symptomatic around the age of 40–60 years, but there can also be multiple lesions, often having a familial characteristic. Most of the CCMs remain asymptomatic throughout life, being detected incidentally following neuroimaging investigations. CCMs causes symptoms ranging from headaches, epilepsy, focal neurological deficit, to extensive life-threatening cerebral hemorrhages [4][11].
Although the vascular lesions, characteristics of CCMs, are frequently located in the central nervous system (CNS), they can often associate lesions in the skin, retina, kidneys, and liver [4][9].
Cutaneous vascular malformations are present in approximately 9% of patients with CCMs, three distinct phenotypes being described as: hyperkeratotic cutaneous capillary -venous malformations (HCCVM) (39%), capillary malformations (CMs) (34%), and venous malformations (VMs) [8][9].
Hyperkeratotic cutaneous capillary—venous malformations (HCCVM, OMIM 116860) are rare cutaneous lesions that occur in a group of patients with CCMs [9]. The cutaneous lesions are congenital and present as thick, irregular, black or purpuric plaques, localized especially on the limbs [4][9][12]. Capillary malformations (CMs) are usually congenital and appear as a port-wine stain or the so-called “punctate” capillary malformation. Venous malformations (VMs) in patients with CCMs can appear as single nodules (often located in one limb) or multiple nodules of variable size [8].
Familial forms are determined by mutations transmitted in an autosomal dominant manner with incomplete penetrance, involving the CCM1 (KRIT1) gene, located on chromosome 7q21–22 [10, the CCM2 (MGC4607 or malcavernin gene) located on chromosome 7p13–15 [13], and the PDCD10 (CCM3) gene located on chromosome 3q25.2–27 [14]. The first mutations identified were a LOF mutations in the KRIT1 gene, which encodes the protein KRIT1 (KREV1 interaction trapped 1), an evolutionarily conserved Ras-family GTPase. So far, it is not precisely known the role of KRIT1 in the formation of cerebral capillaries and veins. The only thing known is that KRIT1 interacts with KREV1/RAP1A, a GTPase of the RAS family. The KRIT1/CCM2/CCM3 complex is known to inhibit MAP3K3. Loss-of-function of KRIT1 and therefore loss of the entire CCM complex leads to activation of MAP3K3 signaling [8].
All the mutations identified in the CCM genes are LOF mutations, which cause a deficiency of the encoded proteins, causing molecular disorganization and dysfunction of endothelial junctions, affecting the maintenance of the integrity of the vascular barrier. KRIT1 (CCM1) mutations were detected most frequently in patients with HCCVM [8][15]. Recently, somatic mutations of the MAP3K3, PIK3CA, MAP2K7 genes have been identified in CCM lesions, which would suggest that in the case of CCMs there may also be somatic mosaicism [16].
2.2. Capillary Malformations (CMs) and RAS/RAF/MAPK/ERK Signaling Pathway
2.2.1. Capillary Malformations (CMs)
Capillary malformations (CMs) are detected in approximately 0.1–2% of newborns [17] and can be isolated (cutaneous CMs) or be part of a complex syndrome. CMs are usually sporadic and appear as flat, red to purple lesions named port-wine stain (nevus flammeus). The etiology of CMs is represented by ASM in the GNAQ gene, located on chromosome 9q21.2 [9][18][19].
Sturge-Weber syndrome (SWS) (OMIM 185300) [9] is a neurocutaneous disorder characterized by capillary malformation (port-wine stains), and choroidal and leptomeningeal vascular malformations most often involving the occipital and posterior parietal lobes [9]. The most common symptoms and signs are facial CM (port-wine stains) typically on the forehead and upper eyelid in the distribution of the 1st and/or 2nd division of the trigeminal nerve, seizures, and glaucoma [9]. Until now, whole genome sequencing (WGS) studies have highlighted the presence of the recurrent somatic mutation c.548G>A (p.R183Q) in the GNAQ (G-alpha q) gene, both in SWS patients and in non -syndromic port-wine stain lesions. The GNAQ p.Arg183Gln mutation that causes the loss of arginine in GNAQ with the reduction of hydrogen bonds between G(q) and GDP is most frequently detected in patients with CMs [18].
In their study, Shirlley et al. [18] identified a nonsynonymous single-nucleotide variant (c.548G→A, p.Arg183Gln) in the GNAQ gene in 88% of patients with SWS and in 92% of patients with non-syndromic CMs (port-wine stains) [18]. They did not identify the respective mutation either in the case of four unrelated patients with cerebrovascular malformation, or in the 6 patients from the control group [18]. The somatic substitutions in GNAQ p.Gln209Leu and GNAQ p.Arg183Gln were detected in patients with uveal melanoma. The most common is the GNAQ p.Gln209Leu mutation which has been shown to overactivate the mitogen-activated protein kinase (MAPK) pathway. The GNAQ p.Arg183Gln mutation has a gain-of-function effect that activates downstream signaling pathways. However, the effect of GNAQ p.Arg183Gln in MAPK signal transduction appears to be weaker in terms of activation of downstream effectors than the effect of the more frequently detected GNAQ p.Gln209Leu substitution in uveal melanoma tissue [18]. Galeffi et al. [19] identified the presence of the GNAQ p.R183Q mutation in most of the patients with SWS that were analyzed, and in one patient, a new GNAQ Q209R mutation was identified [19].
2.2.2. Capillary Malformations (CM)—Arteriovenous Malformation (AVM)
Cutaneous vascular lesions specific to patients with Capillary Malformation (CM)—Arteriovenous Malformation (AVM) (CM-AVM) are small, multifocal, disseminated, round/oval shaped, and pink to red colored macules or papules surrounded by a pale halo on Doppler ultrasound [20]. They are present at birth, but can also appear later in life. CM-AVMs are frequently associated with high-flow arteriovenous malformations (AVMs) or arteriovenous fistulas (AVFs) located in muscles, skin, and other tissues (intracranial, intraspinal), aneurysmal malformation of the vein of Galen or Parkers Weber syndrome (PWS) [20][21][22].
Mutations of the RASA1 gene (RASp21 protein activator 1; OMIM 139150) [9] (located on the chromosome 5q14.3) and transmitted in an autosomal dominant manner, are responsible for different forms of VMs (CMs, AVMs, AVFs), as single manifestations, or in complex combinations, as in the case of PWS [20][22]. Initially, two loci for inherited CMs were identified on chromosome 5 (5q14-21 and 5q13-22), and later, the RASA1 (RAS P21 Protein Activator 1) gene was identified as a candidate gene for atypical CMs with AVMs and AVFs, and sporadically, in PWS [20][22].
The spectrum of capillary malformation—arteriovenous malformation syndrome 1 (CM-AVM1) includes all cases of VMs associated with RASA1 mutations. Later, other studies reported the presence of RASA1 mutation in atypical CMs as well as cases with aneurysmal malformation of the vein of Galen (VOGM) and Hereditary hemorrhagic telangiectasia (HHT) [22].
CM-AVM1 syndrome follows an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity (this aspect may suggest the involvement of a two-hit mechanism). The RASA1 gene encodes a GTPase-activating protein (p120-RasGAP) and is a negative regulator of RAS/MAPK and MAPK/ERK pathways, transforming Ras protein into its inactive form, variations in p120-RasGAP protein level, having an impact on angiogenesis [23]. The active GTP form of Ras interacts with the Raf protein which is responsible for the phosphorylation of proteins involved in cell growth, proliferation, and differentiation. After activation of the receptor tyrosine kinase, p120-RasGAP is recruited to the cell membrane where it inhibits the RAS/MAPK/ERK signaling pathway and regulates cell growth, differentiation, and proliferation. The p120-RasGAP protein interacts with p190RhoGAP or FAK (focal adhesion kinase), both of which have a role in the movement of the vascular endothelium [23].
Unlike CM-AVM1, CM-AVM2 is caused by LOF mutations of the EPHB4 gene, located on chromosome 7q22.1 [24]. The EPHB4 gene encodes the protein EPHB4 (Ephrin type-B receptor 4), a transmembrane tyrosine kinase receptor that binds to ephrin-B2 and plays an essential role in vasculogenesis [9]. EPHB4 inhibits the RAS/MAPK/ERK pathway by interacting with p120-RasGAP. This inhibitory effect is lost in CM-AVM2 leading to a constitutive activation of RAS/MAPK/ERK signaling [4][24].
2.3. Arteriovenous Malformations and RAS/RAF/MAPK/ERK Signaling Pathway
Arteriovenous Malformations (AVMs) can be located in any organ in the body, including visceral or peripheral structures, and in the CNS. AVMs often destroy adjacent structures during their development/growth, being the most aggressive vascular malformations. Most of the time, it is not possible to completely remove the lesion by embolization or surgical intervention, and the remaining lesions cause the worsening of the AVM [4][23].
In their study, Couto et al. [25] identified the presence of a ASM mutation in the MAP2K1 (Mitogen-activated protein kinase kinase 1) gene, located on chromosome 15q22.31) [9] in seven of the ten analyzed patients, who presented peripheral or extracranial AVMs [25]. The ASM mutations in the MAP2K1 gene (which encodes MAP-extracellular signal-regulated kinase 1, MEK1), activate the RAS/MAPK signaling pathway that controls numerous cellular and developmental processes. Somatic mutations in the KRAS (Kirsten rat sarcoma virus) gene, located on chromosome 12p12.1 (OMIM 190070) [9] identified by Couto et al. [25] have also been identified in various neoplasms (including melanoma, lung cancer, and hematopoietic malignancies) and have been shown to constitutively increase MEK1 activity [25]. Somatic mutations that affect the proteins that intervene upstream of MEK1, are detected both in different types of vascular malformations and in different types of cancer [25][26].
Nikolaev et al. [27] detected two ASM mutation (c.35G→A, p.Gly12Asp and c.35G→T, p.Gly12Val), in the KRAS gene, in the case of 45 of the 72 patients with cerebral AVMs [27]. The researchers demonstrated that the expression of the mutant KRAS gene in endothelial cells (ECs) in vitro induces an increase in ERK activity, and an increase in the expression of genes involved in angiogenesis and Notch signaling (Notch homologous protein of the neurogenic locus), as well as increase of cell migration capacity [27]. These processes were reversed by inhibiting MAPK (mitogen-activated protein kinase)–ERK signaling [27].
In another study, Al-Olabi et al. [28] identified mutations in KRAS, BRAF (proto-oncogene B-raf) (localized on chromosome 7q34) and MAP2K1 genes, which supports the role of RAS/RAF/MAPK signaling in AVMs [28].
2.4. Lymphatic Malformations and RAS/RAF/MAPK/ERK Signaling Pathway
Gorham-Stout disease (GSD) (OMIM 123880), which is also known as “vanishing bone disease”, “disappearing bone disease”, or cystic angiomatosis of bone, is a rare disease of massive osteolysis associated with proliferation and overgrowth of lymphatic vessels [9]. GSD belongs to complex lymphatic malformations (LMs) [9] and may affect any bone in the body and can be monostotic or polyostotic. The ribs, spine, pelvis, skull, collarbone (clavicle), and jaw are frequently affected. Symptoms at presentation are dependent upon the location(s) of the disease; the most common symptom is localized pain accompanied by swelling of the affected area and functional impotence. The disease may be discovered after a pathological fracture determined by osteolysis and osteopenia. The etiology of GSD is not fully known, and the anatomopathological data indicate disorganized lymphangiogenesis [9][29].
Homayun-Sepehr et al. [30] identified an ASM mutation (p.G12V) in the KRAS gene in a patient with GSD [30].
Kaposiform lymphangiomatosis (KLA) is a rare, frequently aggressive, systemic disorder of the lymphatic vasculature, characterized by multifocal malformed lymphatic channels, frequently located in the thoracic cavity, but also involving the spleen or the skeleton. Specific clinical manifestations include pericardial and pleural effusions, cough, dyspnea, bleeding, and fractures secondary to bone involvement, with poor prognosis [31].
Barclay et al. [32] identified an ASM in the NRAS gene (c.182A>G, p.Q61R) in 10 of the 11 patients with KLA, suggesting that RAS signaling is important for the development of KLA. The NRAS gene located on chromosome 1p13.2 [9] encodes the N-Ras protein that is involved primarily in regulating cell division [32]. In addition, the activating NRAS p.Q61R variant is known as a hotspot variant, frequently identified in several types of human cancer, especially melanoma [32].
2.5. Vascular Tumors and RAS/RAF/MAPK/ERK Signaling Pathway
2.5.1. Pyogenic Granuloma
Pyogenic granuloma (PG) (also called lobular capillary hemangioma) is an acquired benign vascular hyperplasia, with an etiology that is not fully known. PG occurs frequently in children and young adults, and manifests as a red papular lesion or a solitary, rapidly growing nodule, prone to bleeding from minor trauma and ulceration [4][33].
PG generally occurs in the skin and mucous membranes (face, trunk, oral cavity), but occasionally, it can be located in the gastrointestinal tract or larynx. PG can occur spontaneously or within capillary malformations (CMs). PG occurs frequently during pregnancy, being called “pregnancy tumor”, or associated with certain drugs. In secondary PGs, detected in some CMs, an ASM mutation in the GNAQ gene (p.Arg183Gln) has been described. Because the CM and concomitant PG have the same mutation, the hypothesis is that the PG originates from the underlying CM cells [33].
The pathogenesis of most sporadic PGs and PGs associated with port-wine stains (PWS) is not fully elucidated. Groesser et al. [33] analyzed 10 cases with PGs secondary to a PWS. The researchers identified a BRAF mutation c.1799T>A (p.Val600Glu) in the case of 8 patients and a NRAS mutation c.182A>G (p.Gln61Arg) in one patient [33]. GNAQ mutation c.548G >A was identified in PGs and underlying PWS respectively, indicating that PGs originate from PWS cells [33]. In 25 patients with sporadic PGs, the researchers identified the BRAF c.1799T>A mutation in 3 of the patients, a BRAF c.1391G>A mutation in one patient, and a KRAS c.37G>C mutation in one patient [33]. The researchers concluded that the BRAF c.1799T>A gene mutation has a major role in the pathogenesis of PGs, especially of the secondary CMs, opening the way for deciphering the genetic basis of PGs [33].
Other studies have reported cases of PGs in which somatic mutations common to those detected in colon cancers have been identified, respectively mutations in the BRAF (p.Val600Glu or p.Gly464Glu), KRAS (p.Gly13Arg), GNA14, and HRAS (p.Q61R, p.E49K, p.Q61R and p.G13S) genes [33][34][35].
Relatively recently, cases of oral PGs were reported, in which no BRAF, KRAS, HRAS, NRAS, GNA11, or GNA14 gene mutations were identified, suggesting that although oral PGs shows activation of the MAPK/ERK pathway, the major molecular events are not fully elucidated [36].
2.5.2. Congenital Hemangioma
Congenital hemangioma (CH) is a rare vascular tumor that forms during intrauterine development. CH are different from common infantile hemangiomas (IH) that enlarge rapidly after birth and immunostain for the cell surface marker GLUT1.1. In contrast, CHs are negative for GLUT1.1, and postnatally, the tumor either rapidly involutes (rapidly involuting congenital hemangioma) (RICH) or regresses (non-involuting congenital hemangioma) (NICH) [37][38].
Starting from the premise that CHs are determined by somatic mutations, Ayturk et al. [37] performed mRNA sequencing from affected tissue in a patient with CH. The researchers identified the mosaic missense mutations that alter glutamine at amino acid 209 (pGlu209Leu) (c.626A>T) in the GNAQ or GNA11 genes in all tested samples. These mutations were different from those detected in capillary malformations (CMs). The researchers then looked for the presence of mutations in the genomic DNA. Both GNAQ and GNA11 pGlu209Leu missense variants identified in patients with CHs are also common mutations in uveal melanoma and have been shown to constitutively activate MAPK and/or YAP (Yes-Associated Protein) signaling [37].
References
- Greene, A.K. Vascular anomalies: Current overview of the field. Clin. Plast. Surg. 2011, 38, 1–5.
- International Society For The Study Of Vascular Anomalies ISSVA. Available online: https://www.issva.org/classification (accessed on 29 August 2022).
- Wetzel-Strong, S.E.; Detter, M.R.; Marchuk, D.A. The pathobiology of vascular malformations: Insights from human and model organism genetics. J. Pathol. 2017, 241, 281–293.
- Queisser, A.; Seront, E.; Boon, L.M.; Vikkula, M. Genetic Basis and Therapies for Vascular Anomalies. Circ. Res. 2021, 129, 155–173.
- Nguyen, H.L.; Boon, L.M.; Vikkula, M. Vascular Anomalies Caused by Abnormal Signaling within Endothelial Cells: Targets for Novel Therapies. Semin. Intervent. Radiol. 2017, 34, 233–238.
- Borst, A.J.; Nakano, T.A.; Blei, F.; Adams, D.M.; Duis, J. A Primer on a Comprehensive Genetic Approach to Vascular Anomalies. Front. Pediatr. 2020, 8, 579591.
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059.
- Colmenero, I.; Knöpfel, N. Venous Malformations in Childhood: Clinical, Histopathological and Genetics Update. Dermatopathology 2021, 8, 477–493.
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org (accessed on 30 August 2022).
- Couto, J.A.; Vivero, M.P.; Kozakewich, H.P.; Taghinia, A.H.; Mulliken, J.B.; Warman, M.L.; Greene, A.K. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am. J. Hum. Genet. 2015, 96, 480–486.
- Du, Z.; Zheng, J.; Zhang, Z.; Wang, Y. Review of the endothelial pathogenic mechanism of TIE2-related venous malformation. J. Vasc. Surg. Venous Lymphat. Disord. 2017, 5, 740–748.
- Eerola, I.; Plate, K.H.; Spiegel, R.; Boon, L.M.; Mulliken, J.B.; Vikkula, M. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associ ated with cerebral capillary malformation. Hum. Mol. Genet. 2000, 9, 1351–1355.
- Yang, L.; Wu, J.; Zhang, J. A Novel CCM2 Gene Mutation Associated With Cerebral Cavernous Malformation. Front. Neurol. 2020, 11, 70.
- Cigoli, M.S.; Avemaria, F.; De Benedetti, S.; Gesu, G.P.; Accorsi, L.G.; Parmigiani, S.; Corona, M.F.; Capra, V.; Mosca, A.; Giovannini, S.; et al. PDCD10 gene mutations in multiple cerebral cavernous malformations. PLoS ONE 2014, 9, e110438.
- Hirota, K.; Akagawa, H.; Kikuchi, A.; Oka, H.; Hino, A.; Mitsuyama, T.; Sasaki, T.; Onda, H.; Kawamata, T.; Kasuya, H. KRIT1 mutations in three Japanese pedigrees with hereditary cavernous malformation. Hum. Genome Var. 2016, 3, 16032.
- Weng, J.; Yang, Y.; Song, D.; Huo, R.; Li, H.; Chen, Y.; Nam, Y.; Zhou, Q.; Jiao, Y.; Fu, W.; et al. Somatic MAP3K3 mutation defines a subclass of cerebral cavernous malformation. Am. J. Hum. Genet. 2021, 108, 942–950.
- Antaya, R.; Elston, D.M. Capillary Malformation. Available online: https://emedicine.medscape.com/article/1084479-overview (accessed on 5 September 2022).
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979.
- Galeffi, F.; Snellings, D.A.; Wetzel-Strong, S.E.; Kastelic, N.; Bullock, J.; Gallione, C.J.; North, P.E.; Marchuk, D.A. A novel somatic mutation in GNAQ in a capillary malformation provides insight into molecular pathogenesis. Angiogenesis 2022, 25, 493–502.
- Lapinski, P.E.; Doosti, A.; Salato, V.; North, P.; Burrows, P.E.; King, P.D. Somatic second hit mutation of RASA1 in vascular endothelial cells in capillary malformation-arteriovenous malformation. Eur. J. Med. Genet. 2018, 61, 11–16.
- Brouillard, P.; Vikkula, M. Genetic causes of vascular malformations. Hum. Mol. Genet. 2007, 16, R140–R149.
- Ustaszewski, A.; Janowska-Głowacka, J.; Wołyńska, K.; Pietrzak, A.; Badura-Stronka, M. Genetic syndromes with vascular malformations—Update on molecular background and diagnostics. Arch. Med. Sci. 2020, 17, 965–991.
- Revencu, N.; Boon, L.M.; Mendola, A.; Cordisco, M.R.; Dubois, J.; Clapuyt, P.; Hammer, F.; Amor, D.J.; Irvine, A.D.; Baselga, E.; et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum. Mutat. 2013, 34, 1632–1641.
- Amyere, M.; Revencu, N.; Helaers, R.; Pairet, E.; Baselga, E.; Cordisco, M.R.; Chung, W.K.; Dubois, J.; Lacour, J.P.; Martorell, L.; et al. Germline Loss-of-Function Mutations in EPHB4 Cause a Second Form of Capillary Malformation-Arteriovenous Malformation (CM-AVM2) Deregulating RAS-MAPK Signaling. Circulation 2017, 136, 1037–1048.
- Couto, J.A.; Huang, A.Y.; Konczyk, D.J.; Goss, J.A.; Fishman, S.J.; Mulliken, J.B.; Warman, M.L.; Greene, A.K. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am. J. Hum. Genet. 2017, 100, 546–554.
- Chakraborty, R.; Hampton, O.A.; Shen, X.; Simko, S.J.; Shih, A.; Abhyankar, H.; Lim, K.P.; Covington, K.R.; Trevino, L.; Dewal, N.; et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014, 124, 3007–3015.
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Rezai Jahromi, B.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.R.; et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N. Engl. J. Med. 2018, 378, 250–261.
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508.
- Angelini, A.; Mosele, N.; Pagliarini, E.; Ruggieri, P. Current concepts from diagnosis to management in Gorham-Stout disease: A systematic narrative review of about 350 cases. EFORT Open Rev. 2022, 7, 35–48.
- Homayun-Sepehr, N.; McCarter, A.L.; Helaers, R.; Galant, C.; Boon, L.M.; Brouillard, P.; Vikkula, M.; Dellinger, M.T. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight 2021, 6, e149831.
- Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=464329 (accessed on 5 September 2022).
- Barclay, S.F.; Inman, K.W.; Luks, V.L.; McIntyre, J.B.; Al-Ibraheemi, A.; Church, A.J.; Perez-Atayde, A.R.; Mangray, S.; Jeng, M.; Kreimer, S.R.; et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet. Med. 2019, 21, 1517–1524.
- Groesser, L.; Peterhof, E.; Evert, M.; Landthaler, M.; Berneburg, M.; Hafner, C. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J. Investig. Dermatol. 2016, 136, 481–486.
- Lim, Y.H.; Douglas, S.R.; Ko, C.J.; Antaya, R.J.; McNiff, J.M.; Zhou, J.; Choate, K.A.; Narayan, D. Somatic activating RAS mutations cause vascular tumors including pyogenic granuloma. J. Investig. Dermatol. 2015, 135, 1698–1700.
- Lim, Y.H.; Bacchiocchi, A.; Qiu, J.; Straub, R.; Bruckner, A.; Bercovitch, L.; Narayan, D.; McNiff, J.; Ko, C.; Robinson-Bostom, L.; et al. GNA14 somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. Am. J. Hum. Genet. 2016, 99, 443–450.
- Pereira, T.D.S.F.; de Amorim, L.S.D.; Pereira, N.B.; Vitório, J.G.; Duarte-Andrade, F.F.; Guimarães, L.M.; Diniz, M.G.; Gomes, C.C.; Gomez, R.S. Oral pyogenic granulomas show MAPK/ERK signaling pathway activation, which occurs independently of BRAF, KRAS, HRAS, NRAS, GNA11, and GNA14 mutations. J. Oral. Pathol. Med. 2019, 48, 906–910.
- Ayturk, U.M.; Couto, J.A.; Hann, S.; Mulliken, J.B.; Williams, K.L.; Huang, A.Y.; Fishman, S.J.; Boyd, T.K.; Kozakewich, H.P.W.; Bischoff, J.; et al. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am. J. Hum. Genet. 2016, 98, 1271.
- Funk, T.; Lim, Y.; Kulungowski, A.M.; Prok, L.; Crombleholme, T.M.; Choate, K.; Bruckner, A.L. Symptomatic congenital hemangioma and congenital hemangiomatosis associated with a somatic activating mutation in GNA11. JAMA Dermatol. 2016, 152, 1015–1020.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Revisions:
2 times
(View History)
Update Date:
31 Oct 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No