 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jerome Moreaux | + 2552 word(s) | 2552 | 2020-11-11 09:10:54 | | | |
| 2 | Catherine Yang | + 1 word(s) | 2553 | 2020-11-24 03:35:53 | | |
Video Upload Options
Polycomb group (PcG) proteins are epigenetic regulators involved in cell fate and carcinogenesis. The emerging roles of PcG in PC differentiation and myelomagenesis position them as potential therapeutic targets in multiple myeloma (MM), a cancer that emerges from plasma cells. Plasma cells (PC) are the main effectors of adaptive immunity, responsible for producing antibodies to defend the body against pathogens. They are the result of a complex highly regulated cell differentiation process, taking place in several anatomical locations and involving unique genetic events. Pathologically, PC can undergo tumorigenesis and cause a group of diseases known as plasma cell dyscrasias, including multiple MM, a severe disease with poor prognosis characterized by the accumulation of malignant PC within the bone marrow, and high clinical and molecular heterogeneity. MM patients frequently develop resistance to treatment, leading to relapse. Below, the roles of PcG proteins in normal and malignant plasma cells and their therapeutic implications are presented.
CC1. Introduction
Since the end of the 19th century, plasma cells (PC) have been a major topic of research in order to understand their function and origin [1]. The hypothesis of PC lymphocytic genesis was first formulated in 1902 by Alexander Maximow in his unitarian theory of hematopoiesis [2]. Then, in 1947, Astrid Fagraeus demonstrated in vitro that PC are the antibody-secreting cells [3]. However, it was not until 1965 that Max Cooper and Robert Good demonstrated that PC derived from lymphocytes in the bursa of Fabricius (by definition B lymphocytes), found in all modern birds (Neorthis) [4].
It is now established that PC constitute the terminal stage of B lymphocyte differentiation and are the major players of the humoral immune response. Under pathological conditions, PC are at the center of allergic and autoimmune hypersensitivity reactions. Moreover, multiple myeloma (MM), a frequent hematologic cancer that in most cases remains untreated, is caused by malignant PC transformation and accumulation in the bone marrow [5]. In this context, many groups are investigating the modulation of the physiological differentiation of PC by epigenetic factors as well as their tumoral transformation.
Polycomb group (PcG) proteins are major epigenetic regulators of gene expression during development and cell fate choice, which, beyond their general role in biology, have gained strong interest for their specific implication in hematological cell differentiation and diseases. The first PcG component, the Polycomb gene, was discovered by Pamela Lewis in Drosophila melanogaster in 1947 [6]. A paradigm establishes that PcGs act as transcriptional repressors, although more recent observations have suggested that PcG might potentiate transcription. The two main PcG complexes are named polycomb repressive complex 1 (PRC1) and polycomb repressive complex 2 (PRC2), and function as multiprotein complexes that display strong evolutionary conservation [7].
2. PcG Complexes
PRC1 is composed of a core that includes the E3 ubiquitin ligase enzymes RING1A or RING1B, and one of the PCGF1-6 subunits. RING1 is the catalytic subunit that catalyzes the monoubiquitylation of lysine 119 of histone H2A (H2AK119ub1) on chromatin and interacts in a mutually exclusive manner with a chromobox protein (CBX 2, 4, 6–8) or RYBP (or its close homolog YAF2). On this basis, mammalian PRC1 complexes that include a CBX subunit have been classified as canonical PRC1 (cPRC1), and PRC1 complexes containing RYBP or YAF2 have been classified as non-canonical PRC1 (ncPRC) [7]. Moreover, depending on the PCGF subunit associated with RING1A/B, different PRC1 complexes have been described and divided into canonical and non-canonical groups (also known as variants) [8] (Figure 1).
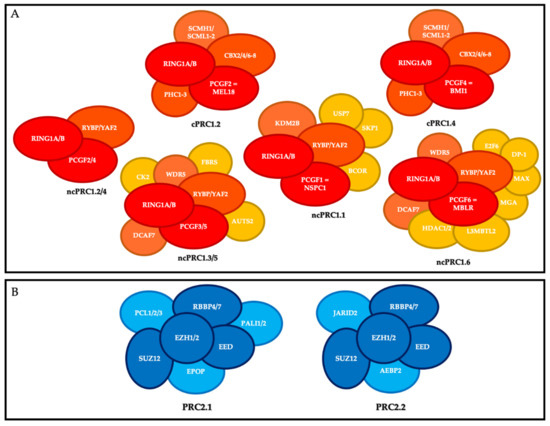
Figure 1. Polycomb repressive complexes (PRC). (A) Composition of canonical PRC1 (cPRC1) and non-canonical PRC1 (ncPRC1). Red, core members; orange, members that define the different canonical and non-canonical complexes; yellow, accessory factors. (B) Composition of PRC2. Dark blue, core members; light blue, members that define the different complexes.
The canonical PRC1s (cPRC1s) are cPRC1.2 and cPRC1.4. In addition to RING1A or RING1B, their core contains MEL18 (PCGF2) and BMI-1 (PCGF4), respectively; one of the CBX2/4/6–8 proteins, which harbor the chromodomain allowing cPRC1 to recognize tri-methylation of lysine 27 of histone H3 (H3K27me3); and one of the three proteins PHC1-3 [9]. cPRC1 also includes the following accessory non-stoichiometric members: SCMH1, and SCMHL1/2 [10].
The non-canonical PRC1s (ncPRC1s) are ncPRC1.1, ncPRC1.2/4, ncPRC1.3/5, and ncPRC1.6. In addition to RING1 subunit, their cores include NSPC1 (PCGF1), PCGF2/4, PCGF3/5, and MBLR (PCGF6), respectively, and RYBP or YAF2. The ncPRC1 group includes many accessory members, particularly KDM2B and BCOR for ncPRC1.1; AUTS2 for PRC1.3/5; and HDAC1/2, E2F6, MAX and MGA for PRC1.6 [10].
PRC2 is composed of a core that includes the histone methyl transferases EZH1 or EZH2, which catalyze methylation of histone H3 at lysine 27 (H3K27me3) on chromatin via its SET domain, as well as its partners EED, SUZ12, and RBBP4/7, which are essential for its function. Depending on the members associated with this core, there are two main PRC2s: PRC2.1 (which includes EPOP, PALI1/2, and PCL1-3) and PRC2.2 (which includes AEBP2 and JARID2) [11].
One of the important points in the biology of PcG proteins is that none of the core members of PRC1 or PRC2 can recognize specific DNA sequences on their own, and therefore they all need to be recruited by partners to regulate the specific expression of their target genes [8]. These partners include accessory proteins that bind unmethylated CG-rich sequences, histone marks, transcription factors, and RNAs, and much remains to be learnt about the precise mechanisms, cell type, and time-specificity of PcG recruitment at their targets [10][12][13] (Figure 2).
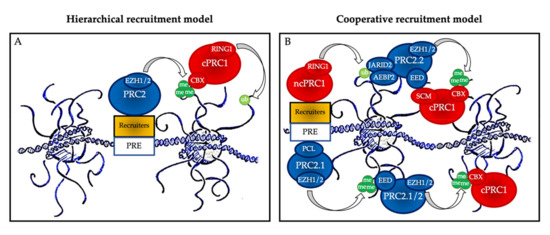
Figure 2. Polycomb group protein chromatin recruitment models. (A) Hierarchical recruitment model: PRC2 is recruited first and deposits H3K27me3 on chromatin via its catalytical subunit EZH1 or EZH2; then, canonical PRC1 (cPRC1) is recruited by a chromobox member CBX on the H3K27me3 mark and deposits H2AK119ub1 on chromatin via its catalytical subunit RING1. (B) Cooperative recruitment model: ncPRC1 complexes deposit H2AK119ub, which recruits PRC2.2 via its JARID2 and AEBP2 subunits. In parallel, PRC2.1 is recruited to unmethylated CpG island DNA via its PCL subunits. PRC2.1 and PRC2.2 complexes deposit H3K27me3, and this mark recruits both more copies of PRC2 and cPRC1. Mutual interactions between the core PRC2 member EED and the cPRC1 member SCM further stabilize their recruitment. PRE: polycomb responsive element (considered as CpG islands in mammals).
The historical hierarchical model described by Wang et al. in 2004 postulates that PRC2 is first recruited to chromatin and deposits H3K27me3. This epigenetic mark is then recognized by the chromodomain of CBX2/4/6–8, allowing recruitment of cPRC1 and the subsequent deposition of H2AK119ub1. According to this model, PRC1 is recruited in a PRC2-dependent manner. More recent data have allowed this model to be considerably refined. We now know that ncPRC1 binds independently on PRC2, either via proteins such as KDM2B (PRC1.1) [12], via interactions with ncRNAs such as Xist (PRC1.3 and PRC1.5) [14], or via transcription factors (TFs) [15]. ncPRC1 complexes deposit H2AK119ub, which recruits PRC2.2 via its JARID2 and AEBP2 subunits [16][17][18]. In parallel, PRC2.1 is recruited to unmethylated CpG island DNA via its PCL subunits [19]. Finally, PRC2 complexes deposit H3K27me3, and this mark recruits both more copies of PRC2 and cPRC1 [11]. Mutual interactions between these proteins further stabilize their recruitment. Therefore, a series of DNA-protein, RNA-protein, protein-protein, and protein-histone interactions lead to stable PcG protein recruitment to their target sites (Figure 2B). In this scenario, multiple signals, including PcG subunit, TF, or ncRNA abundance as well as CpG density and length can modulate PcG recruitment, offering multiple possibilities for regulation, leading to maintenance, stabilization, or displacement, depending on regulatory cues.
3. Lymphopoiesis and Plasma Cell Differentiation
The production of high affinity immunoglobulins (Igs; antibodies) is the critical point of the humoral immune response and the principle of vaccination strategies [20]. Antibody-secreting cells (ASC) include pre-plasmablasts (prePB), plasmablasts (PB), and PC (i.e., the final stages of B lymphocyte differentiation) [21].
B cell lymphopoiesis consists of several steps that take place in different anatomical compartments. Briefly, during the early medullary phase, hematopoietic stem cells (HSC) and lymphoid progenitors successively generate pro-B lymphocytes, pre-B lymphocytes, and immature B lymphocytes, independently of the antigen. During this phase, VDJ recombination of Ig heavy and light chains and selection of functional B lymphocytes take place [14][22]. Subsequently, immature B lymphocytes leave the medullary compartment to reach the spleen where, during a transitional phase, they become naive mature B lymphocytes that express IgM- and IgD-positive B cell receptors (BCRs) [23][24].
Naïve mature B lymphocytes have an immuno-surveillance role and begin to circulate in the follicles of the various secondary lymphoid organs (lymph nodes, spleen, and mucosa-associated lymphoid tissue). In these organs, the first contact between a naive B lymphocyte and its antigen triggers the primary humoral immune response. At this stage, a distinction is made between thymus-dependent (TD) antigens, which require the intervention of a follicular helper T lymphocyte (TFH, or more strictly a pre-TFH) to fully activate the mature B lymphocyte, and thymus-independent (TI) antigens, which can directly activate mature B cells by stimulating the toll-like receptor (TLR) pathway (TI type I antigens) or by cross-linking large numbers of BCRs simultaneously (TI type II antigens) [25].
The interaction between a mature B lymphocyte (through its BCR) and its specific antigen triggers two phenomena grouped under the term of “BCR triggering”: internalization of the BCR–antigen complex, and signal transduction through the BCR pathway. The BCR–antigen complex is internalized by a clathrin-dependent mechanism called receptor-mediated endocytosis and is driven to the endosomal pathway, where the antigen is loaded onto an MHC class II molecule that is then returned to the plasma membrane. In this process, the B lymphocyte assumes the role of professional antigen-presenting cell and becomes able to interact with a pre-TFH lymphocyte. The BCR pathway includes the Lyn and Syk tyrosine kinases that induce the expression of co-stimulatory molecules and chemotactic receptors necessary for subsequent events [5][26].
At this stage, the B lymphocyte is not yet fully activated, but only pre-activated (primed), and can also be defined as B lymphoblast [27]. Upon contact with the antigen and activation of the BCR pathway, the naive mature B lymphocyte exits its quiescent state (reversible G0) and enters the G1 phase of the cell cycle, where it remains paused, awaiting an additional stimulus to complete its activation and proliferate [5]. This additional mitogenic stimulus can come from two different sources, depending on the nature of the antigen that triggered the pre-activation. TD antigens must interact with a pre-TFH cell, whereas TI antigens have intrinsic activating activity [5]. Contact with pre-TFH cells occurs in the B-T interface zone of secondary lymphoid organs through a structure known as the immune synapse, a specialized cell-cell junction organized in a bull’s eye pattern that was first described by Kupfer and his collaborators in 1998 [28] and reviewed by Dustin in 2015 [29].
Following this activation, the B lymphoblast can choose between the follicular response and the extra-follicular response (Figure 3). The extra-follicular response is the only response to TI antigens and constitutes the early response to TD antigens. It allows the rapid production of short-lived ASC (PB and then PC) that secrete IgM with low affinity for the antigen [30].
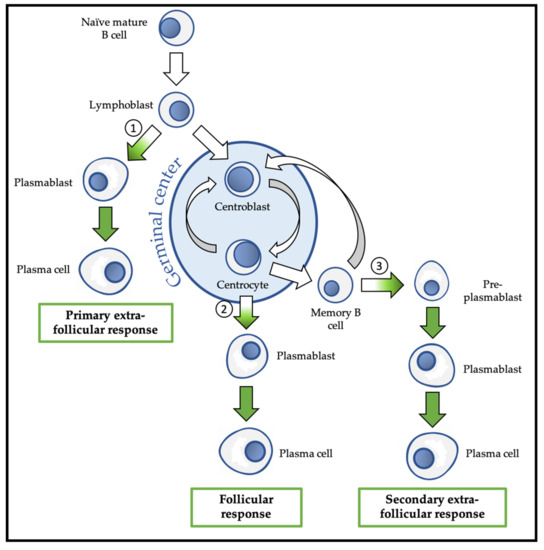
Figure 3. Overview of physiological differentiation of plasma cells (PC). Upon primary antigen contact, naïve mature B cells are activated and become lymphoblasts that can choose between two pathways: the primary extra-follicular response and the primary follicular response. In the primary extra-follicular response, lymphoblasts immediately differentiate into plasmablasts (PB) then into short-lived PC and produce immunoglobin M (IgM). In the primary follicular response, lymphoblasts enter a lymphoid follicle and form a germinal centre, while differentiating into centroblasts and then centrocytes. During the germinal centre reaction, immunoglobulin class switch recombination (CSR) and somatic hypermutation (SHM) take place. After selection, the centrocytes differentiate either into memory B cells, or into PB and then PC that produce IgG, IgA, or IgE. Upon secondary antigen contact, memory B cells are activated and can choose between a secondary extra-follicular response and a secondary follicular response. The secondary follicular response is similar to the primary follicular response and follows the same steps. In the secondary extra-follicular response, memory B cells immediately differentiate into pre-plasmablasts (pre-PB), PB, and then PC. The numbers indicate the cell fate decision points, where the antibody-secreting cell (ASC) program is switched on. Green arrows indicate cell differentiation steps where the ASC program takes place.
The follicular response is specific to TD antigens and involves the formation of a secondary lymphoid follicle in which the processes of somatic hypermutation (SHM) and isotypic switching/class switch recombination (CSR) are carried out to generate antibodies with up to 30,000 times more affinity for the antigen and with a downstream isotype (IgG, IgA, or IgE) with new effector functions, respectively. The follicular response induces the differentiation of activated B lymphocytes into centroblasts and centrocytes, followed by the generation of PB that differentiate into short-lived (SLPC) and long-lived (LLPC) PC. LLPC can survive in specialized niches for decades [31]. The follicular response also generates memory B cells (MBC) [32][33].
MBC are the main players in the secondary humoral response that is triggered by a new contact with the same antigen. MBC activation can lead to their engagement in the extra-follicular response during which they differentiate into pre-PB, PB, and finally PC, or in a new follicular response with SHM, and possibly CSR [5].
Despite these various cell differentiation pathways, terminal B cell differentiation is transcriptionally regulated at two main developmental stages, each involving a specific network of transcriptional factors: the guardian network of the B phenotype, which includes PAX5, BACH2, and BCL6, and the vector network of the ASC phenotype, which includes PRDM1 (BLIMP1), IRF4, and XBP1 [30][20]. The complexity and beauty of PC differentiation lies in the regulation of the decision point between these two mutually exclusive networks that requires epigenetic mechanisms and PcG proteins.
The other critical point of the follicular response is the regulation of the genetic events that target the loci of the Ig genes during the germinal center reaction, namely, SHM and CSR, the understanding of which remains one of the main challenges in humoral immunology and pathological hematology. These two processes are orchestrated by the same enzyme, AID, a thymidine deaminase of the APOBEC family that exerts a physiological mutagenic activity exclusively in developing B lymphocytes [34][35]. PcG proteins have also been implicated in the regulation of AID-catalyzed reactions [36].
4. Multiple Myeloma
Multiple myeloma (MM) is a malignant hemopathy that affected nearly 230,000 people worldwide in 2015, with an incidence of around 130,000 new cases per year [37]. It is the second most frequent malignant hemopathy (10–13% in 2017) after non-Hodgkin lymphoma, and accounted for 1.7% of all cancers and 2% of all cancer deaths in 2017. MM is observed mainly in >50-year-old adults, and the average age at diagnosis is between 63 and 70 years.
MM development (i.e., myelomagenesis) is a multi-step process characterized by the appearance of genomic alterations and microenvironmental changes [38][39].
MM is a highly heterogeneous disease at the molecular and clinical levels [40][41][42][43]. Epigenetic modifications including DNA methylation, chromatin accessibility, and histone modifications have been reported in MM in association with pathogenic impact [44][45][46][47][48]. Moreover, the many epigenetic alterations observed in MM contribute to this biological heterogeneity and also to treatment resistance [49]. Due to their reversible nature, epigenetic alterations are particularly interesting for new targeted therapy strategies in MM [50][51]. PcG proteins are the subject of growing interest in MM with the hope of improving therapeutic management of MM patients.
References
- Ribatti, D. The discovery of plasma cells: An historical note. Immunol. Lett. 2017, 188, 64–67.
- Maximow, A.A. Chapter III-C. In Experimentelle Untersuchungen über Entzündliche Neubildung von Bindegewebe; Fischer: Leipzig, Germany, 1902.
- Fagraeus, A. Plasma Cellular Reaction and its Relation to the Formation of Antibodies in vitro. Nature 1947, 159, 499.
- Cooper, M.D.; Peterson, R.D.A.; Good, R.A. Delineation of the thymic and bursal lymphoid systems in chicken. Nature 1965, 205, 103–146.
- Cyster, J.G.; Allen, C.D.C. B cell responses—Cell interaction dynamics and decisions. Cell 2019, 177, 524–540.
- Lewis, P.H. Melanogaster-New mutants: Report of Pamela H. Lewis. Dros. Inform. Serv. 1947, 21, 69.
- Chittock, E.C.; Latwiel, S.; Miller, T.C.R.; Müller, C.W. Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 2017, 45, 193–205.
- Blackledge, N.P.; Rose, N.R.; Klose, R.J. Targeting polycomb systems to regulate gene expression: Modifications to a complex story. Nat. Rev. Mol. Cell Biol. 2015, 16, 643–649.
- Ma, R.; Zhang, Y.; Sun, T.; Cheng, B. Epigenetic regulation by polycomb group complexes: Focus on roles of CBX proteins. J. Zhejiang Univ. Sci. B 2014, 15, 412–428.
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57.
- Healy, E.; Mucha, M.; Glancy, E.; Fitzpatrick, D.J.; Conway, E.; Neikes, H.K.; Monger, C.; Van Mierlo, G.; Baltissen, M.P.; Koseki, Y.; et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76, 437–452.e6.
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459.
- Simon, J.A.; Kingston, R.E. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 2013, 49, 808–824.
- Mårtensson, I.-L.; Almqvist, N.; Grimsholm, O.; Bernardi, A.I. The pre-B cell receptor checkpoint. FEBS Lett. 2010, 584, 2572–2579.
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.-T.; Kathrein, K.L.; Woo, A.J.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct Recruitment of Polycomb Repressive Complex 1 (PRC1) to Chromatin by Core Binding Transcription Factors. Mol. Cell 2012, 45, 330–343.
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571.
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661.
- Tamburri, S.; Lavarone, E.; Fernández-Pérez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e5.
- Perino, M.; van Mierlo, G.; Loh, C.; Wardle, S.M.T.; Zijlmans, D.W.; Marks, H.; Veenstra, G.J.C. Two Functional Axes of Feedback-Enforced PRC2 Recruitment in Mouse Embryonic Stem Cells. Stem Cell Rep. 2020.
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242.
- Tellier, J.; Nutt, S.L. Plasma cells: The programming of an antibody-secreting machine. Eur. J. Immunol. 2019, 49, 30–37.
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580.
- Loder, B.F.; Mutschler, B.; Ray, R.J.; Paige, C.J.; Sideras, P.; Torres, R.; Lamers, M.C.; Carsetti, R. B Cell Development in the Spleen Takes Place in Discrete Steps and Is Determined by the Quality of B Cell Receptor–Derived Signals. J. Exp. Med. 1999, 190, 75–90.
- Chung, J.B.; Silverman, M.; Monroe, J.G. Transitional B cells: Step by step towards immune competence. Trends Immunol. 2003, 24, 342–348.
- Liu, Y.-J.; Zhang, J.; Lane, P.J.L.; Chan, E.Y.-T.; Maclennan, I.C.M. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur. J. Immunol. 1991, 21, 2951–2962.
- Kurosaki, T.; Shinohara, H.; Baba, Y. B Cell Signaling and Fate Decision. Annu. Rev. Immunol. 2010, 28, 21–55.
- Murphy, K.; Weaver, C. Chapter 1-18: Lymphocytes activated by antigen proliferate in the peripheral lymphoid organs, generating effector cells and immunological memory. In Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2016.
- Kupfer, A.; Singer, S.J. The specific interaction of helper T cells and antigen-presenting B cells. IV. Membrane and cytoskeletal reorganizations in the bound T cell as a function of antigen dose. J. Exp. Med. 1989, 170, 1697–1713.
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033.
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171.
- Nguyen, D.C.; Joyner, C.J.; Sanz, I.; Lee, F.E.-H. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 2019, 10.
- Shlomchik, M.J.; Weisel, F. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 2012, 247, 52–63.
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol 2008, 8, 22–33.
- Muramatsu, M.; Sankaranand, V.S.; Anant, S.; Sugai, M.; Kinoshita, K.; Davidson, N.O.; Honjo, T. Specific Expression of Activation-induced Cytidine Deaminase (AID), a Novel Member of the RNA-editing Deaminase Family in Germinal Center B Cells. J. Biol. Chem. 1999, 274, 18470–18476.
- Xu, Z.; Pone, E.J.; Al-Qahtani, A.; Park, S.-R.; Zan, H.; Casali, P. Regulation of aicda expression and AID activity: Relevance to somatic hypermutation and class switch DNA recombination. Crit. Rev. Immunol. 2007, 27, 367–397.
- Caganova, M.; Carrisi, C.; Varano, G.; Mainoldi, F.; Zanardi, F.; Germain, P.-L.; George, L.; Alberghini, F.; Ferrarini, L.; Talukder, A.K.; et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J. Clin. Investig. 2013, 123, 5009–5022.
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma. JAMA Oncol. 2018, 4, 1221–1227.
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187.
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10.
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65.
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028.
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553.
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101.
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562.
- Kaiser, M.F.; Johnson, D.C.; Wu, P.; Walker, B.A.; Brioli, A.; Mirabella, F.; Wardell, C.P.; Melchor, L.; Davies, F.E.; Morgan, G.J. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 2013, 122, 219–226.
- Ordoñez, R.; Kulis, M.; Russiñol, N.; Chapaprieta, V.; Carrasco-Leon, A.; García-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227.
- Agirre, X.; Castellano, G.; Pascual, M.; Heath, S.; Segura, V.; Bergmann, A.; Esteve, A.; Merkel, A.; Raineri, E.; Agueda, L.; et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015, 34, 478–487.
- Jin, Y.; Chen, K.; De Paepe, A.; Hellqvist, E.; Krstic, A.D.; Metang, L.; Gustafsson, C.; Davis, R.E.; Levy, Y.M.; Surapaneni, R.; et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018, 131, 2138–2150.
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115.
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front. Oncol. 2018, 8.
- Dupéré-Richer, D.; Licht, J.D. Epigenetic Regulatory Mutations and Epigenetic Therapy for Multiple Myeloma. Curr. Opin. Hematol. 2017, 24, 336–344.

