 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Soumya Basu | -- | 11800 | 2022-10-28 07:30:14 | | | |
| 2 | Jason Zhu | -244 word(s) | 11556 | 2022-10-31 03:27:29 | | | | |
| 3 | Soumya Basu | Meta information modification | 11556 | 2022-11-14 06:42:29 | | |
Video Upload Options
Peroxisome proliferator-activated receptor-γ (PPAR-γ) has emerged as one of the most extensively studied transcription factors since its discovery in 1990, highlighting its importance in the etiology and treatment of numerous diseases involving various types of cancer, type 2 diabetes mellitus, autoimmune, dermatological and cardiovascular disorders. Ligands are regarded as the key determinant for the tissue-specific activation of PPAR-γ. However, the mechanism governing this process is merely a contradictory debate which is yet to be systematically researched. Either these receptors get weakly activated by endogenous or natural ligands or leads to a direct over-activation process by synthetic ligands, serving as complete full agonists. Therefore, fine-tuning on the action of PPAR-γ and more subtle modulation can be a rewarding approach which might open new avenues for the treatment of several diseases.
1. Introduction
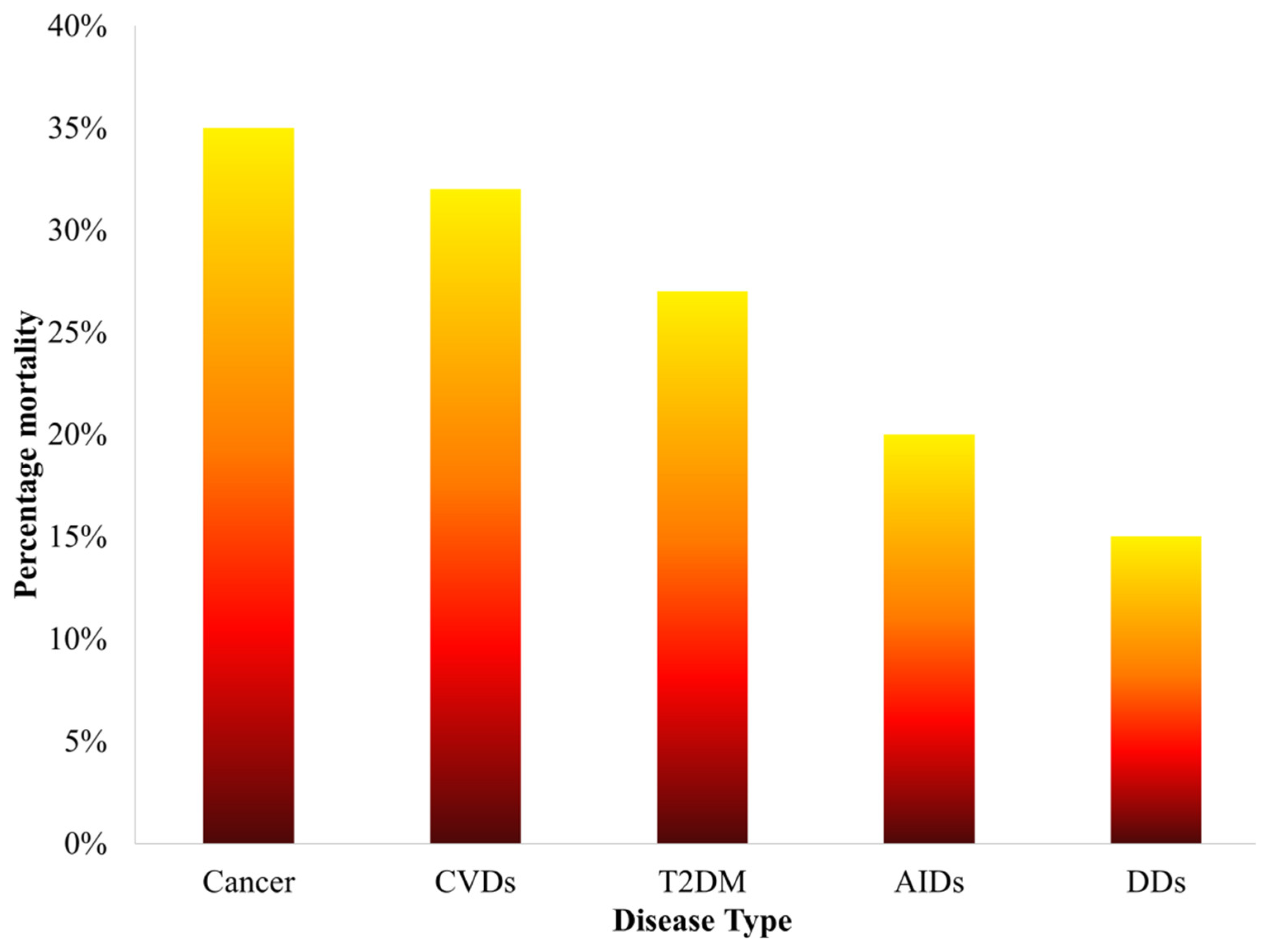
Figure 1. Statistics of percentage mortality caused by various diseases over the past five years.
Table 1. Overview of the basic metabolic function of PPAR isotypes.
|
PPAR Isotype |
Site of elevated expression |
Cellular mechanisms initiated |
Biological function |
Genes targeted |
References |
|
PPAR-α |
Heart, liver and kidney |
β-oxidation of fatty acids, synthesis of lipoprotein and regulating metabolic pathways of amino acids |
Complementation of metabolic reaction to fasting |
Hydroxymethylglutaryl CoA synthase 2 (HMG- CoA S2) |
[15] |
|
PPAR-β/δ |
Adipocytes and macrophages |
Differentiation pathways of adipocytes and production of triglycerides |
Differentiation pathways of adipocytes and fatty acid trapping |
Fatty acid binding protein 4 (FABP4) |
[16] |
|
PPAR-γ |
Adipocytes, skin and brain |
β-oxidation of fatty acids |
Coordination of muscle fibers and determination of its types |
Acyl CoA Oxidase (AOX) |
[17] |
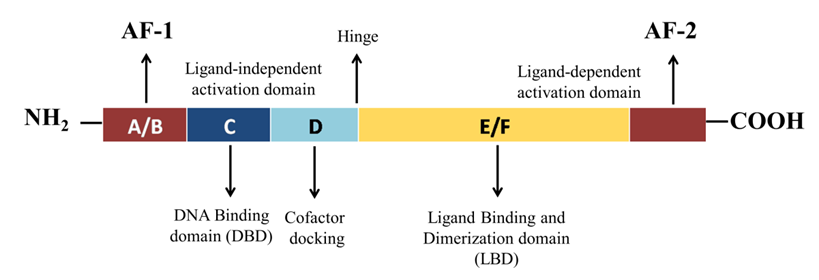
2. Structural Mechanism of PPAR-γ
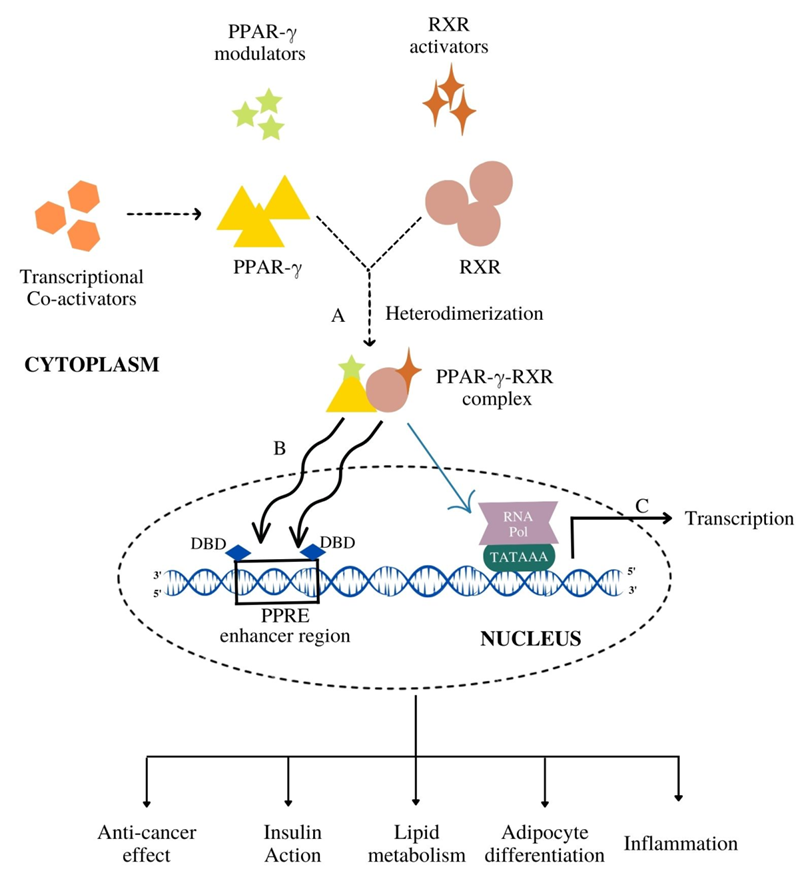
3. Functional Diversity of PPAR-γ
3.1. Anti-Cancer Effect
3.2. Insulin Action
3.3. Lipid Metabolism
3.4. Adipocyte Differentiation
3.5. Inflammation
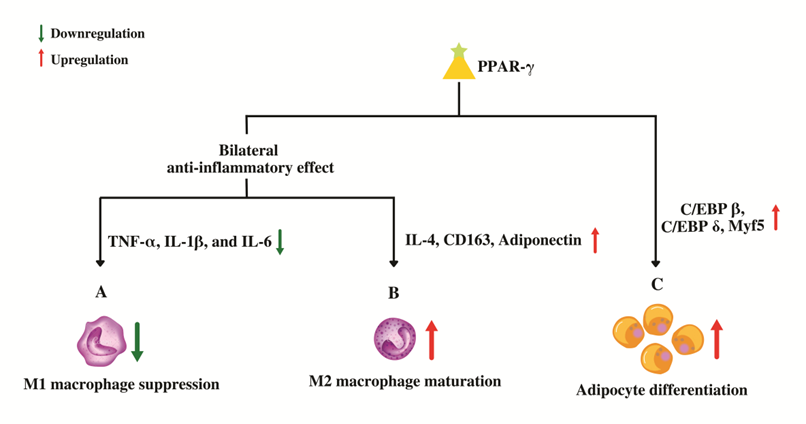
Figure 4. Role of PPAR-γ in macrophage conversion and adipocyte differentiation. (A) PPAR-γ suppresses the genes that code for pro-inflammatory molecules which in-turn prevents the maturation of pro-inflammatory wild-type "M1" macrophages. (B) PPAR-γ promotes the maturation of anti-inflammatory "M2" macrophages by up-regulating anti-inflammatory genes leading to an overall bilateral anti-inflammatory effect. (C) PPAR-γ regulates adipocyte differentiation by upregulating the genes involved in proliferative stage of terminal differentiation.
By enhancing tight junction proteins, PPAR-γ has been also shown to defend the blood–brain barrier’s (BBB) integrity. Through the measurement of NF-κB transcriptional activity and FITC-Dextran permeability, Talé and colleagues evaluated the impact of the PPAR-γ agonist, pioglitazone on markers of the inflammatory response and permeability changes brought on by hyperglycemia [64]. Their results indicated that high glucose-mediated activation of NF-κB is blocked by PPAR-γ agonist. Additionally, PPAR-γ agonists guard against FITC-Dextran permeability elevation that is aggravated by hyperglycemic conditions viz. T2DM. This shows that PPAR-γ has a protective function against the inflammatory and permeability changes that are caused by hyperglycemia in endothelial cells. It is noteworthy that mild PPAR-γ activators such as telmisartan, amorfrutin, and other PPAR-γ partial agonists significantly defend against PPAR-γ gene expression downregulation brought on by hyperglycemia [64].
4. PPAR-γ Activation by Various Ligands
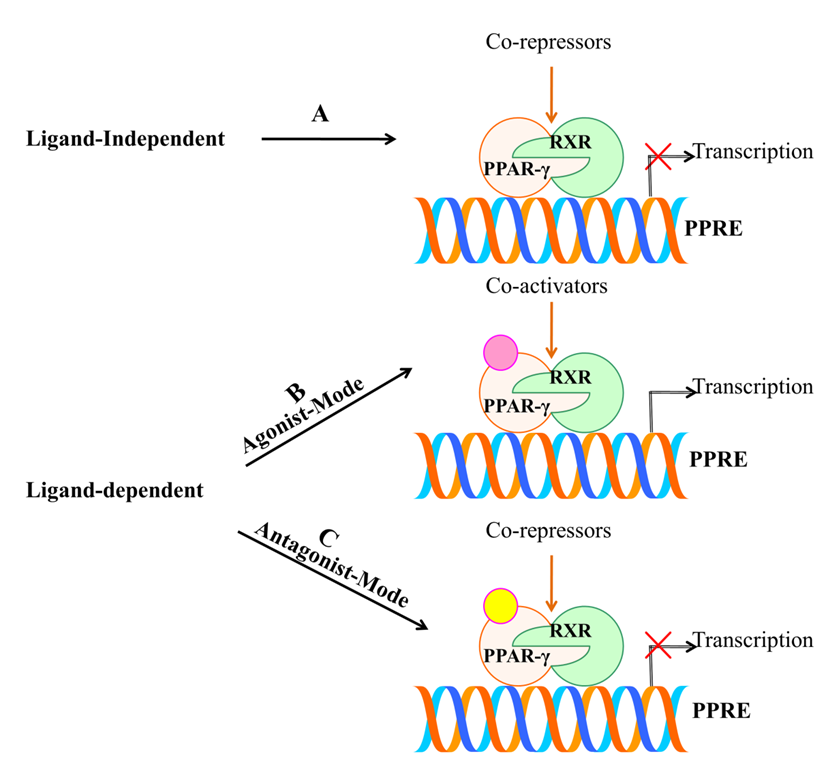
4.1. Endogenous Ligands
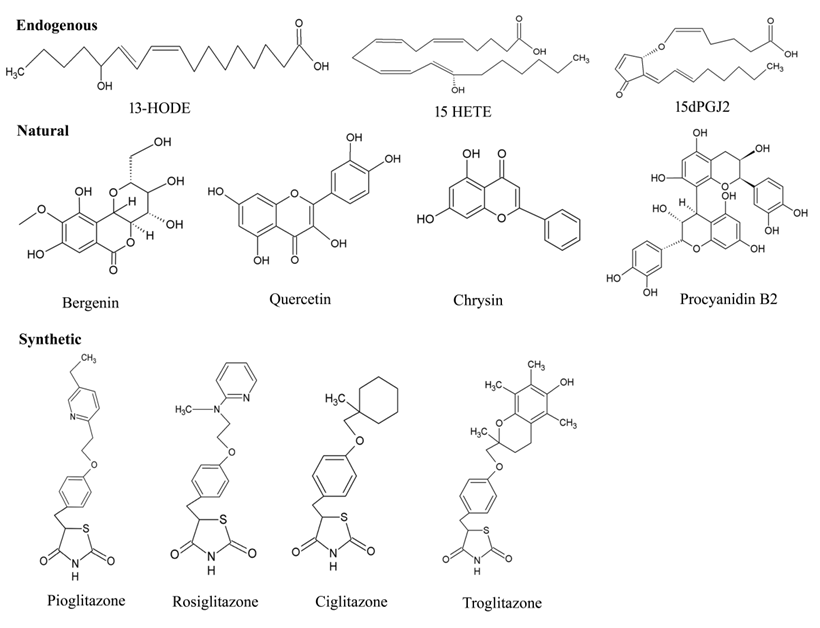
Figure 6. Chemical structures of various types PPAR-γ ligands (Endogenous, natural, and synthetic).
4.2. Natural Ligands
4.3. Synthetic Ligands
5. PPAR-γ Agonists in Various Diseases
5.1. Cancer
5.2. Cardiovascular Disease (CVDs)
5.3. Type 2 Diabetes Mellitus (T2DM)
5.4. Autoimmune Diseases (AIDs)
5.5. Inflammatory Diseases
5.6. Dermatological Diseases (DDs)
Table 2 List of ligands activating PPAR-γ and their significant role in various diseases.
|
Type of PPAR-γ ligand |
Name of the Ligand |
Source |
Disease |
Effect in Disease |
References |
|
|
Endogeneous |
13-HODE |
n-3 LC-PUFA |
Cancer |
Anti-proliferative activity, cell cycle arrest (G1) and apoptosis |
[157] |
|
|
Multiple sclerosis |
Reduced clinical severity of allergic encephalomyelitis |
[158] |
|
|||
|
15-HETE |
n-3 LC-PUFA |
Cancer |
Anti-proliferative activity, cell cycle arrest (G1), Apoptosis |
[159] |
|
|
|
CVD |
Anti-platelet and anti-thrombotic effects |
[160] |
|
|||
|
15d-PGJ2 |
Prostaglandin J2 derivative |
Cancer |
Cell cycle arrest, apoptosis and reducing ornithine decarboxylase activity |
[161]
|
|
|
|
Inflammatory disorders |
Regulates expression of surface proteins, T-cell activation, and related inflammatory cytokines |
[162] |
|
|||
|
AID |
Anti-inflammatory effects in primary biliary cirrhosis patients |
[141] |
|
|||
|
|
|
|
Asthma |
Inhibited T(H)2 type cytokine IL-5production |
[163] |
|
|
Natural |
Procyanidin B2 |
Flavonoid |
Hepatic diseases |
Inhibited nicotine-induced pyroptosis |
[164] |
|
|
Artepillin C |
Baccharisdracunculifolia |
T2DM |
Induced adipocyte differentiation and glucose uptake |
[165] |
|
|
|
Lectins and viscotoxins |
Herbs-Viscum album L. |
Cancer |
apoptosis, inhibition of angiogenesis |
[166] |
|
|
|
Bergenin |
Herb of Saxifragastolonifera Curt. |
Inflammatory disorders |
alleviated disease symptoms of dextran sulfate sodium (DSS)-induced colitis |
[81] |
|
|
|
|
|
Asthma |
Prevented GLS1-dependent glutaminolysis |
[167] |
|
|
|
Antioxidants (Ascorbic acid and phytochemicals) |
Whole-apple extracts |
Cancer |
Inhibition of tumor-cell proliferation in prostate and breast cancer |
[82] |
|
|
|
1,1-Bis(3′-indolyl)-1-(p-trifluoromethylphenyl)methane |
p-substituted phenyl analogues |
Cancer |
Cell cycle arrest (G0/G1-S) in endometrial cancer |
[168] |
|
|
|
Chrysin |
Flavonoid |
Asthma |
Alleviated ovalbumin-airway hyperresponsiveness |
[169] |
|
|
|
Quercetin |
Flavonoid |
Cancer |
Tumor-inhibitory effects in breast cancer |
[170] |
|
|
|
Cancer |
Anti-proliferative and anti-migratory effects in lung cancer |
[171] |
|
|||
|
|
||||||
|
CucurbitaneTriterpenoid |
Extract of wild bitter gourd (Momordicacharantia) |
Cancer |
Anti-proliferative effect induced apoptotic death in breast cancer cells |
[172] |
|
|
|
Insulin resistance |
Induced adipocyte differentiation and glucose uptake |
[173] |
|
|||
|
T2DM |
Induced glucose uptake |
[174] |
|
|||
|
Methanolic extractPterocarpusmarsupium |
isoflavone |
T2DM |
Induced glucose uptake and elevated Glut-4 |
[175] |
|
|
|
Synthetic |
Pioglitazone |
TZD |
Neurological disease |
Inhibited mTOR activation and prevented increase of IL‑1β and IL‑6. |
[176] |
|
|
Neurological disease |
Reduced hyperalgesia and astrocyte activation |
[177] |
|
|||
|
Psoriatic Arthritis Response |
Inhibited angiogenesis and suppressed pro-inflammatory cytokines |
[178] |
|
|||
|
Asthma |
Reduced regulator of G protein 4 |
[179] |
|
|||
|
SRD |
Inhibited bleomycin-induced skin fibrosis |
[98] |
|
|||
|
Rosiglitazone |
TZD |
Ischemia Stroke |
Limited postischemic injury in normal and diabetic hearts |
[180] |
|
|
|
Asthma |
Reduced bronchial inflammation |
[181] |
|
|||
|
Allergy |
Decreased ROS generation, expression of T(H)2 cell cytokines in lungs after ovalbumin inhalation |
[182] |
|
|||
|
Ciglitazone |
TZD |
Cancer |
Inhibitory effects on lung cancer |
[183] |
|
|
|
Cancer |
Inhibitory effects on prostrate cancer |
[184] |
|
|||
|
Troglitazone
|
TZD |
Cancer |
Reduced c-Myc levels in prostate cancer
|
[185] |
|
|
|
|
|
Psoriasis |
Inhibited proliferation of psoriatic human keratinocytes |
[186] |
|
|
|
GW347845 |
Non-TZD |
AID |
Anti-inflammatory and anti-proliferative effects |
[138] |
|
|
* 13-HODE-13-Hydroxyoctadecadienoic acid; n-3 LC-PUFA-n-3 long chain polyunsaturated fatty acids; 15-HETE-15-hydroxyeicosatetraenoic acid; 15d-PGJ2-15-Deoxy-∆-12,14-Prostaglandin J2; T2DM, Type 2 diabetes mellitus; TZD, Thiazolidinedione.
6. Challenges Faced and Knowledge Gaps for PPAR-γ Agonists
7. Significance of PPAR-γ Partial Agonist
8. PPAR-γ Partial Agonists Involved in Post-Transcriptional Modification and Disease-Fate Decision
8.1. Phosphorylation
8.2. SUMOylation
8.3. Ubiquitination
Table 3. Effect of PPAR-γ partial agonists in disease fate decision.
|
PPAR-γ partial agonist |
Type of compound |
Disease |
Effect in Disease |
References |
|
SPPAR-γM5 |
SPPAR-γM |
T2DM
|
Reduced the insulin resistance index |
[69] |
|
PAR-1622 |
SPPAR-γM |
T2DM |
Induced adipocyte differentiation and improved hyperglycemia |
[214] |
|
PAM-1616 |
SPPAR-γM |
T2DM |
Improvedhyperglycemia |
[223] |
|
FK614 |
SPPAR-γM |
T2DM |
Reduced the insulin resistance index |
[224] |
|
F12016 |
SPPAR-γM |
T2DM |
Insulin-sensitizing and glucose-lowering properties |
[132] |
|
KDT501 |
Chemically derived from substituted 1,3-cyclopentadione |
Inflammatory disorders |
Anti-inflammatory effects in monocytes/macrophages |
[225] |
|
GQ-16 |
TZD-Derived |
Obesity |
Reduced high fat diet-induced weight gain |
[211] |
|
|
|
Cancer |
Anti-proliferative effects in breast cancer |
[212] |
| Telmisartan |
Angiotensin type 1 receptor blocker |
Inflammatory disorders |
cerebroprotective effect |
[213] |
|
T2DM |
Ameliorated vascular endothelial dysfunction and protected against diabetic vascular complications |
[202] |
SPPAR-γM-selective PPAR-γ modulators; PAR-1622-(S)-2-ethoxy-3(4-(5-(4-(5-(methoxymethyl)isoxazol-3-yl)phenyl)-3-methylthiophen-2-yl)methoxy)phenyl)propanoic Acid; PAM-1616-(S)-2-ethoxy-3-(4-((3-methyl-5-(4-(3-methylisoxazol-5-yl) phenyl) thiophen-2-yl) methoxy) phenyl) propanoic acid; FK614-3-(2,4-dichlorobenzyl)-2-methyl-N-(pentylsulfonyl)-3-Hbenzimidazole-5-carboxamide; F12016-2-[2-(1,2-dimethyl-1H-indol-3-yl)-2-oxo-acetylamino]-benzamide; KDT501-Potassium salt of the n-(isobutyl) congener of a tetrahydro iso-α acid; GQ-16-(5Z)-5-(5-bromo-2-methoxy-benzylidene)-3-(4-methyl-benzyl)-thiazolidine-2,4-dione.
9. PPAR-γ Partial Agonists in Cancer Therapeutics
Table 4. List of PPAR-γ partial agonists and their significant role in cancer therapeutics.
|
PPAR-γ partial agonist |
Type of compound |
Effect in Disease |
Cell line |
Type of cancer |
References |
|
Deoxyelephantopin |
Natural |
Apoptosis and cell cycle arrest (G(2)/M) |
HeLa |
Cervix |
[69] |
|
Halofenate
|
SPPAR-γM
|
Anti-proliferative effects |
MM96L |
Melanoma |
[226] |
|
Tetrazanbigen |
Sterol isoquinoline derivative |
Anti-proliferative effects |
HepG2 and A549 |
Liver and lung |
[220] |
|
HydroxyCinnamic Acid Derivatives |
p-coumaric acid and ferulic acid |
Anti-proliferative effects |
K562
|
Chronic Myeloid Leukemia
|
[222] |
|
Telmisartan |
Angiotensin II (Ang II) receptor blocker |
Apoptosis and anti-proliferative effects |
Caki-1,T24, LNCaP, PC3, DU-145 and NEC-8 |
Renal, bladder, prostate and testicular |
[203] |
|
|
|
Anti-proliferative effects |
A549 |
Lung |
[227] |
10. PPAR-γ Partial Agonists under Clinical Trials
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33.
- Hu, M.; Wang, X.; Tan, J.; Yang, J.; Gao, X.; Yang, Y. Causal Associations between Paternal Longevity and Risks of Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 233.
- Knudsen, J.S.; Knudsen, S.S.; Hulman, A.; Witte, D.R.; Gregg, E.W.; Lauritzen, T.; Pedersen, L.; Sørensen, H.T.; Thomsen, R.W. Changes in type 2 diabetes incidence and mortality associated with introduction of HbA1c as diagnostic option: A Danish 24-year population-based study. Lancet Reg. Health-Eur. 2022, 14, 100291.
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 2020, 12, e8094.
- Quinto, R.M.; Iani, L.; Vincenzo, F.D.; Russo, F.; Porcelli, P.; Abeni, D. Does Guided Written Disclosure Reduce Distress and Improve Psychological Functioning in Patients with Skin Diseases? Int. J. Environ. Res. Public Health 2022, 19, 2943.
- Colca, J.R. The TZD insulin sensitizer clue provides a new route into diabetes drug discovery. Expert Opin. Drug Discov. 2015, 10, 1259–1270.
- Yuan, Z.; Luo, G.; Li, X.; Chen, J.; Wu, J.; Peng, Y. PPAR-γ inhibits HMGB1 expression through upregulation of miR-142-3p In Vitro and In Vivo. Cell. Signal. 2016, 28, 158–164.
- Asakawa, M.; Takano, H.; Nagai, T.; Uozumi, H.; Hasegawa, H.; Kubota, N.; Saito, T.; Masuda, Y.; Kadowaki, T.; Komuro, I. Peroxisome Proliferator-Activated Receptor Plays a Critical Role in Inhibition of Cardiac Hypertrophy In Vitro and In Vivo. Circulation 2002, 105, 1240–1246.
- Tyagi, S.; Gupta, P.; Saini, A.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236.
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892.
- Youssef, J.; Badr, M.Z. PPARs: History and Advances. Methods Mol. Biol. 2013, 952, 1–6.
- Decara, J.; Rivera, P.; López-Gambero, A.J.; Serrano, A.; Pavón, F.J.; Baixeras, E.; de Fonseca, F.R.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730.
- Rosen, E.D.; Spiegelman, B.M. PPAR: A Nuclear Regulator of Metabolism, Differentiation, and Cell Growth. J. Biol. Chem. 2001, 276, 37731–37734.
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep. 2016, 6, 34792.
- Han, L.; Shen, W.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 2017, 13, 259–278.
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-α and PPAR-β/δ on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS ONE 2018, 13, e0191365.
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Up-regulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-γ. Cell. Physiol. Biochem. 2018, 48, 1317–1331.
- Plutzky, J. The PPAR-RXR Transcriptional Complex in the Vasculature. Circ. Res. 2011, 108, 1002–1016.
- Garcia-Bates, T.M.; Lehmann, G.M.; Simpson-Haidaris, P.J.; Bernstein, S.H.; Sime, P.J.; Phipps, R.P. Role of Peroxisome Proliferator-Activated Receptor Gamma and Its Ligands in the Treatment of Hematological Malignancies. PPAR Res. 2008, 2008, 834612.
- Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activated receptors and cancers: Complex stories. Nat. Rev. Cancer 2004, 4, 61–70.
- Grommes, C.; Landreth, G.L.; Heneka, M.T. Antineoplastic effects of peroxisome proliferatoractivated receptor gamma agonists. Lancet Oncol. 2004, 5, 419–429.
- Bennett, R.G. Ligand-Independent Coactivation of Peroxisome Proliferator-Activated Receptor Gamma; Springer: Cham, Switherland, 2021; pp. 519–535.
- Cronet, P.; Petersen, J.F.; Folmer, R.; Blomberg, N.; Sjöblom, K.; Karlsson, U.; Lindstedt, E.; Bamberg, K. Structure of the PPAR-alpha and -gamma Ligand Binding Domain in Complex with AZ 242 Ligand Selectivity and Agonist Activation in the PPAR Family. Structure 2001, 9, 699–706.
- Yu, C.; Markan, K.; Temple, K.A.; Deplewski, D.; Brady, M.J.; Cohen, R.N. The Nuclear Receptor Co-repressors NCoR and SMRT Decrease Peroxisome Proliferator-activated Receptor gamma Transcriptional Activity and Repress 3T3-L1 Adipogenesis. J. Biol. Chem. 2005, 280, 13600–13605.
- Emont, M.P.; Mantis, S.; Kahn, J.H.; Landeche, M.; Han, X.; Sargis, R.M.; Cohen, R.N. Silencing Mediator of Retinoid and Thyroid Hormone Receptors (SMRT) regulates glucocorticoid action in adipocytes. Mol. Cell. Endocrinol. 2015, 407, 52–56.
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-γ–RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356.
- Bugge, A.; Grøntved, L.; Aagaard, M.M.; Borup, R.; Mandrup, S. The PPARgamma2 A/B-Domain Plays a Gene-Specific Role in Transactivation and Cofactor Recruitment. Mol. Endocrinol. 2009, 23, 794–808.
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234.
- Lehrke, M.; Lazar, M.A. The Many Faces of PPAR. Cell 2005, 123, 993–999.
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553.
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112.
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8, 31.
- Augimeri, G.; Bonofiglio, D. PPARgamma: A Potential Intrinsic and Extrinsic Molecular Target for Breast Cancer Therapy. Biomedicines 2021, 9, 543.
- Wagner, N.; Wagner, K. Peroxisome Proliferator-Activated Receptors and the Hallmarks of Cancer. Cells 2022, 11, 2432.
- Wagner, N.; Wagner, K. PPARs and Angiogenesis—Implications in Pathology. Int. J. Mol. Sci. 2020, 21, 5723.
- Basilotta, R.; Lanza, M.; Casili, G.; Chisari, G.; Munao, S.; Colarossi, L.; Cucinotta, L.; Campolo, M.; Esposito, E.; Paterniti, I. Potential Therapeutic Effects of PPAR Ligands in Glioblastoma. Cells 2022, 11, 621.
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal–Epithelial Crosstalk and Carcinogenesis. Cancers 2021, 13, 2153.
- Yang, F.; Zhang, Z.; Xin, D.; Shi, C.; Wu, J.; Guo, Y.; Guan, Y. Peroxisome proliferator-activated receptor gamma ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines. Acta Pharmacol. Sin. 2005, 26, 753–761.
- Bruin, J.E.; Petrik, J.J.; Hyslop, J.R.; Raha, S.; Tarnopolsky, M.A.; Gerstein, H.C.; Holloway, A.C. Rosiglitazone improves pancreatic mitochondrial function in an animal model of dysglycemia: Role of the insulin-like growth factor axis. Endocrine 2010, 37, 303–311.
- Vijayababu, M.R.; Arunkumar, A.; Kanagaraj, P.; Arunakaran, J. Effects of quercetin on insulin-like growth factors (IGFs) and their binding protein-3 (IGFBP-3) secretion and induction of apoptosis in human prostate cancer cells. J. Carcinog. 2006, 5, 10.
- Babcook, M.A.; Gupta, S. Apigenin modulates insulin-like growth factor axis: Implicationsfor prevention and therapy of prostate cancer. Curr. Drug Targets 2012. Online ahead of print.
- Camp, H.S.; Tafuri, S.R. Regulation of Peroxisome Proliferator-activated Receptor Activity by Mitogen-activated Protein Kinase. J. Biol. Chem. 1997, 272, 10811–10816.
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2022, 83, 556–569.
- Wood, W.M.; Sharma, V.; Bauerle, K.T.; Pike, L.A.; Zhou, Q.; Fretwell, D.L.; Schweppe, R.E.; Haugen, B.R. PPARγ Promotes Growth and Invasion of Thyroid Cancer Cells. PPAR Res. 2011, 2011, 171765.
- Mueller, E.; Smith, M.; Sarraf, P.; Kroll, T.; Aiyer, A.; Kaufman, D.S.; Oh, W.; Demetri, G.; Figg, W.D.; Zhou, X.; et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 10990–10995.
- Gianì, F.; Vella, V.; Nicolosi, M.L.; Fierabracci, A.; Lotta, S.; Malaguarnera, R.; Belfiore, A.; Vigneri, R.; Frasca, F. Thyrospheres From Normal or Malignant Thyroid Tissue Have Different Biological, Functional, and Genetic Features. J. Clin. Endocrinol. Metab. 2015, 100, E1168–E1178.
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822.
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Alex, G.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358.
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPARγ and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 818945.
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3611–3616.
- Phua, W.; Wong, M.; Liao, Z.; Tan, N. An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2018, 19, 1425.
- Choi, S.; Park, J.; Choi, J.H. Revisiting PPARγ as a target for the treatment of metabolic disorders. BMB Rep. 2014, 47, 599–608.
- Qaoud, M.T.; Almasri, I.; Önkol, T. Peroxisome Proliferator-Activated Receptors as Superior Targets for Treating Diabetic Disease, Design Strategies-Review Article. Turk. J. Pharm. Sci. 2022, 19, 353–370.
- Quinn, C.E.; Hamilton, P.K.; Lockhart, C.J.; McVeigh, G.E. Thiazolidinediones: Effects on insulin resistance and the cardiovascular system. Br. J. Pharmacol. 2008, 153, 636–645.
- Saltiel, A.R.; Olefsky, J.M. Thiazolidinediones in the Treatment of Insulin Resistance and Type II Diabetes. Diabetes 1996, 45, 1661–1669.
- Blaschke, F.; Takata, Y.; Caglayan, E.; Law, R.E.; Hsueh, W.A. Obesity, Peroxisome Proliferator-Activated Receptor, and Atherosclerosis in Type 2 Diabetes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 28–40.
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–22.
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215.
- Gesta, S.; Tseng, Y.; Kahn, C.R. Developmental Origin of Fat: Tracking Obesity to Its Source. Cell 2007, 131, 242–256.
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717.
- Wu, H.; Li, X.; Shen, C. Peroxisome Proliferator-Activated Receptor in White and Brown Adipocyte Regulation and Differentiation. Physiol. Res. 2020, 67, 759–773.
- Chu, D.; Tao, Y. Human thermogenic adipocytes: A reflection on types of adipocyte, developmental origin, and potential application. J. Physiol. Biochem. 2016, 73, 1–4.
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.; Gao, Y.; Bennett, M.V.; et al. Peroxisome proliferator-activated receptorγ (PPARγ): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163–164, 27–58.
- Chi, T.; Wang, M.; Wang, X.; Yang, K.; Xie, F.; Liao, Z.; Wei, P. PPARγ Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 737776.
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17.
- Mu, F.; Jing, Y.; Ning, B.; Huang, J.; Cui, T.; Guo, Y.; You, X.; Yan, X.; Li, H.; Wang, N. Peroxisome proliferator-activated receptor isoforms differentially regulate preadipocyte proliferation, apoptosis, and differentiation in chickens. Poult. Sci. 2020, 99, 6410–6421.
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial Agonists Activate PPARgamma Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271.
- Ahsan, W. The Journey of Thiazolidinediones as Modulators of PPARs for the Management of Diabetes: A Current Perspective. Curr. Pharm. Des. 2019, 25, 2540–2554.
- Zou, G.; Gao, Z.; Wang, J.; Zhang, Y.; Ding, H.; Huang, J.; Chen, L.; Guo, Y.; Jiang, H.; Shen, X. Deoxyelephantopin inhibits cancer cell proliferation and functions as a selective partial agonist against PPARgamma. Biochem. Pharmacol. 2008, 75, 1381–1392.
- Zhang, H.; Zhou, R.; Li, L.; Chen, J.; Chen, L.; Li, C.; Ding, H.; Yu, L.; Hu, L.; Jiang, H.; et al. Danthron Functions as a Retinoic X Receptor Antagonist by Stabilizing Tetramers of the Receptor. J. Biol. Chem. 2011, 286, 1868–1875.
- Ammazzalorso, A.; DeFilippis, B.; Giampietro, L.; Amoroso, R. Blocking the Peroxisome Proliferator-Activated Receptor (PPAR): An Overview. ChemMedChem 2013, 8, 1609–1616.
- Li, Y.; Yin, Z.; Dong, Y.; Wang, S.; Monroig, Ó.; Tocher, D.R.; You, C. Pparγ Is Involved in the Transcriptional Regulation of Liver LC-PUFA Biosynthesis by Targeting the Δ6Δ5 Fatty Acyl Desaturase Gene in the Marine Teleost Siganus canaliculatus. Mar. Biotechnol. 2018, 21, 19–29.
- Jara-Gutiérrez, Á.; Baladrón, V. The Role of Prostaglandins in Different Types of Cancer. Cells 2021, 10, 1487.
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163.
- Lathion, C.; Michalik, L.; Wahli, W. Physiological ligands of PPARs in inflammation and lipid homeostasis. Future Lipidol. 2006, 1, 191–201.
- Marcone, S.; Evans, P.; Fitzgerald, D.J. 15-Deoxy-Δ12,14-Prostaglandin J2 Modifies Components of the Proteasome and Inhibits Inflammatory Responses in Human Endothelial Cells. Front. Immunol. 2016, 7, 459.
- Zhao, X.; Zhang, Y.; Strong, R.; Grotta, J.C.; Aronowski, J. 15d-Prostaglandin J2 Activates Peroxisome Proliferator-Activated Receptor-gamma, Promotes Expression of Catalase, and Reduces Inflammation, Behavioral Dysfunction, and Neuronal Loss after Intracerebral Hemorrhage in Rats. J. Cereb. Blood Flow Metab. 2005, 26, 811–820.
- Vangaveti, V.; Shashidhar, V.; Collier, F.; Hodge, J.; Rush, C.; Malabu, U.; Baune, B.; Kennedy, R.L. 9- and 13-HODE regulate fatty acid binding protein-4 in human macrophages, but does not involve HODE/GPR132 axis in PPAR-γ regulation of FABP4. Ther. Adv. Endocrinol. Metab. 2018, 9, 137–150.
- Campbell, S.E.; Stone, W.L.; Whaley, S.G.; Qui, M.; Krishnan, K. Gamma (gamma) tocopherol up-regulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer 2003, 3, 25.
- Afzal, S.; Sattar, M.A.; Johns, E.J.; Eseyin, O.A.; Attiq, A. Antioxidant Potential of Adiponectin and Full PPAR-γ Agonist in Correcting Streptozotocin-Induced Vascular Abnormality in Spontaneously Hypertensive Rats. PPAR Res. 2021, 2021, 6661181.
- Wang, K.; Li, Y.; Lv, Q.; Li, X.; Dai, Y.; Wei, Z. Bergenin, Acting as an Agonist of PPARgamma, Ameliorates Experimental Colitis in Mice through Improving Expression of SIRT1, and Therefore Inhibiting NF-B-Mediated Macrophage Activation. Front. Pharmacol. 2018, 8, 981.
- Reagan-Shaw, S.; Eggert, D.; Mukhtar, H.; Ahmad, N. Antiproliferative Effects of Apple Peel Extract Against Cancer Cells. Nutr. Cancer 2010, 62, 517–524.
- Pferschy-Wenzig, E.; Atanasov, A.G.; Malainer, C.; Noha, S.M.; Kunert, O.; Schuster, D.; Heiss, E.H.; Oberlies, N.H.; Wagner, H.; Bauer, R.; et al. Identification of Isosilybin A from Milk Thistle Seeds as an Agonist of Peroxisome Proliferator-Activated Receptor Gamma. J. Nat. Prod. 2014, 77, 842–847.
- Ma, S.; Huang, Y.; Zhao, Y.; Du, G.; Feng, L.; Huang, C.; Li, Y.; Guo, F. Prenylflavone derivatives from the seeds of Psoralea corylifolia exhibited PPAR-γ agonist activity. Phytochem. Lett. 2016, 16, 213–218.
- Weidner, C.; Wowro, S.J.; Rousseau, M.; Freiwald, A.; Kodelja, V.; Abdel-Aziz, H.; Kelber, O.; Sauer, S. Antidiabetic Effects of Chamomile Flowers Extract in Obese Mice through Transcriptional Stimulation of Nutrient Sensors of the Peroxisome Proliferator-Activated Receptor (PPAR) Family. PLoS ONE 2013, 8, e80335.
- Hiben, M.G.; Haan, L.d.; Spenkelink, B.; Wesseling, S.; Vervoort, J.; Rietjens, I.M.C.M. Induction of peroxisome proliferator activated receptorγ (PPARγ) mediated gene expression and inhibition of induced nitric oxide production by Maerua subcordata (Gilg) DeWolf. BMC Complement. Med. Ther. 2020, 20, 80.
- Chuang, C.; Yeh, C.; Yeh, S.; Lin, E.; Wang, L.; Wang, Y. Quercetin metabolites inhibit MMP-2 expression in A549 lung cancer cells by PPAR-γ associated mechanisms. J. Nutr. Biochem. 2016, 33, 45–53.
- Han, X.; Liu, C.; Gao, N.; Zhao, J.; Xu, J. RETRACTED: Kaempferol suppresses proliferation but increases apoptosis and autophagy by up-regulating microRNA-340 in human lung cancer cells. Biomed. Pharmacother. 2018, 108, 809–816.
- Rauf, A.; Imran, M.; Khan, I.A.; ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130.
- Zhong, Y.; Krisanapun, C.; Lee, S.; Nualsanit, T.; Sams, C.; Peungvicha, P.; Baek, S.J. Molecular targets of apigenin in colorectal cancer cells: Involvement of p21, NAG-1 and p53. Eur. J. Cancer 2010, 46, 3365–3374.
- Dumasia, R.; Eagle, K.; Kline-Rogers, E.; May, N.; Cho, L.; Mukherjee, D. Role of PPAR-γ Agonist Thiazolidinediones in Treatment of Pre-Diabetic and Diabetic Individuals: A Cardiovascular Perspective. Curr. Drug Target-Cardiovasc. Hematol. Disord. 2005, 5, 377–386.
- Wu, Y.; Sreeharsha, N.; Sharma, S.; Mishra, A.; Singh, A.K.; Gubbiyappa, S.K. Anticancer Effect of Rosiglitazone, a PPAR-γ Agonist against Diethylnitrosamine-Induced Lung Carcinogenesis. ACS Omega 2020, 5, 5334–5339.
- Li, M.; Lee, T.W.; Yim, A.P.; Mok, T.S.; Chen, G.G. Apoptosis induced by troglitazone is both peroxisome proliferator-activated receptor-γ and ERK-dependent in human non-small lung cancer cells. J. Cell. Physiol. 2006, 209, 428–438.
- Lewis, J.D.; Ferrara, A.; Peng, T.; Hedderson, M.; Bilker, W.B.; Quesenberry, C.P.; Vaughn, D.J.; Nessel, L.; Selby, J.; Strom, B.L. Risk of Bladder Cancer Among Diabetic Patients Treated With Pioglitazone. Diabetes Care 2011, 34, 916–922.
- Cellai, I.; Benvenuti, S.; Luciani, P.; Galli, A.; Ceni, E.; Simi, L.; Baglioni, S.; Muratori, M.; Ottanelli, B.; Serio, M.; et al. Antineoplastic effects of rosiglitazone and PPARgamma transactivation in neuroblastoma cells. Br. J. Cancer 2006, 95, 879–888.
- Xu, X.; Wang, J.; Jiang, H.; Meng, L.; Lang, B. Rosiglitazone induces apoptosis on human bladder cancer 5637 and T24 cell lines. Int. J. Clin. Exp. Pathol. 2017, 10, 10197–10204.
- Mody, M.; Dharker, N.; Bloomston, M.; Wang, P.; Chou, F.; Glickman, T.S.; McCaffrey, T.; Yang, Z.; Pumfery, A.; Lee, D.; et al. Rosiglitazone sensitizes MDA-MB-231 breast cancer cells to anti-tumor effects of tumor necrosis factor-, CH11 and CYC202. Endocr.-Relat. Cancer 2007, 14, 305–315.
- Kanemaru, M.; Asai, J.; Jo, J.; Arita, T.; Kawai-Ohnishi, M.; Tsutsumi, M.; Wada, M.; Tabata, Y.; Katoh, N. Nanoparticle-mediated local delivery of pioglitazone attenuates bleomycin-induced skin fibrosis. J. Dermatol. Sci. 2019, 93, 41–49.
- Zhang, H.-L.; Zhang, Z.-X.; Xu, Y.-J. Ciglitazone inhibits growth of lung cancer cells A549 In Vitro and In Vivo: An experimental study. Zhonghua Zhong Liu Za Zhi 2004, 26, 531–534.
- Hawcroft, G.; Gardner, S.H.; Hull, M.A. Activation of Peroxisome Proliferator-Activated Receptor gamma Does Not Explain the Antiproliferative Activity of the Nonsteroidal Anti-Inflammatory Drug Indomethacin on Human Colorectal Cancer Cells. J. Pharmacol. Exp. Ther. 2003, 305, 632–637.
- Puhl, A.C.; Milton, F.A.; Cvoro, A.; Sieglaff, D.H.; Campos, J.C.; Bernardes, A.; Filgueira, C.S.; Lindemann, J.L.; Deng, T.; Neves, F.A.; et al. Mechanisms of Peroxisome Proliferator Activated Receptor Regulation by Non-steroidal Anti-inflammatory Drugs. Nucl. Recept. Signal. 2015, 13, e004.
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Zhang, J.; Liu, J.; Reshetov, I.V.; et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front. Oncol. 2022, 12, 820968.
- Liu, Y.; Chen, C.; Wang, X.; Sun, Y.; Zhang, J.; Chen, J.; Shi, Y. An Epigenetic Role of Mitochondria in Cancer. Cells 2022, 11, 2518.
- Li, J. 15-Deoxy-∆-12,14-Prostaglandin J2 (15d-PGJ2), an Endogenous Ligand of PPAR-γ Function and Mechanism. PPAR Res. 2019, 2019, 7242030.
- Toaldo, C.; Pizzimenti, S.; Cerbone, A.; Pettazzoni, P.; Menegatti, E.; Daniela, B.; Minelli, R.; Giglioni, B.; Dianzani, M.U.; Ferretti, C.; et al. PPAR ligands inhibit telomerase activity and hTERT expression through modulation of the Myc/Mad/Max network in colon cancer cells. J. Cell. Mol. Med. 2009, 14, 1347–1357.
- Siavash, H.; Nikitakis, N.G.; Sauk, J.J. Abrogation of IL-6-mediated JAK signalling by the cyclopentenone prostaglandin 15d-PGJ2 in oral squamous carcinoma cells. Br. J. Cancer 2004, 91, 1074–1080.
- Chen, Y. 15d-PGJ2 inhibits cell growth and induces apoptosis of MCG-803 human gastric cancer cell line. World J. Gastroenterol. 2003, 9, 2149.
- Tate, T.; Xiang, T.; Wobker, S.E.; Zhou, M.; Chen, X.; Kim, H.; Batourina, E.; Lin, C.; Kim, W.Y.; Lu, C.; et al. Pparg signaling controls bladder cancer subtype and immune exclusion. Nat. Commun. 2021, 12, 6160.
- Augimeri, G.; Giordano, C.; Gelsomino, L.; Plastina, P.; Barone, I.; Catalano, S.; Andò, S.; Bonofiglio, D. The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies. Cancers 2020, 12, 2623.
- Dwyer-Nield, L.D.; McArthur, D.G.; Hudish, T.M.; Hudish, L.I.; Mirita, C.; Sompel, K.; Smith, A.J.; Alavi, K.; Ghosh, M.; Merrick, D.T.; et al. PPARgamma agonism inhibits progression of premalignant lesions in a murine lung squamous cell carcinoma model. Int. J. Cancer 2022. Online ahead of print.
- Zhang, H.; You, L.; Zhao, M. Rosiglitazone attenuates paraquat-induced lung fibrosis in rats in a PPAR gamma-dependent manner. Eur. J. Pharmacol. 2019, 851, 133–143.
- Cao, L.; Chen, X.; Wang, Q.; Huang, X.; Zhen, M.; Zhang, L.; Li, W.; Bi, J. Upregulation of PTEN involved in rosiglitazone-induced apoptosis in human hepatocellular carcinoma cells. Acta Pharmacol. Sin. 2007, 28, 879–887.
- Higuchi, T.; Yamamoto, J.; Sugisawa, N.; Tashiro, Y.; Nishino, H.; Yamamoto, N.; Hayashi, K.; Kimura, H.; Miwa, S.; Igarashi, K.; et al. PPAR Agonist Pioglitazone in Combination With Cisplatinum Arrests a Chemotherapy-resistant Osteosarcoma PDOX Model. Cancer Genom.-Proteom. 2019, 17, 35–40.
- Cheon, C.W.; Kim, D.H.; Kim, D.H.; Cho, Y.H.; Kim, J.H. Effects of ciglitazone and troglitazone on the proliferation of human stomach cancer cells. World J. Gastroenterol. 2009, 15, 310.
- Adorni, R.; Zanatta, F.; D’Addario, M.; Atella, F.; Costantino, E.; Iaderosa, C.; Petarle, G.; Steca, P. Health-Related Lifestyle Profiles in Healthy Adults: Associations with Sociodemographic Indicators, Dispositional Optimism, and Sense of Coherence. Nutrients 2021, 13, 3778.
- Pasqua, T.; Rocca, C.; Giglio, A.; Angelone, T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. J. Clin. Med. 2021, 10, 721.
- Yan, L.; Zhang, J.D.; Wang, B.; Lv, Y.J.; Jiang, H.; Liu, G.L.; Qiao, Y.; Ren, M.; Guo, X.F. Quercetin Inhibits Left Ventricular Hypertrophy in Spontaneously Hypertensive Rats and Inhibits Angiotensin II-Induced H9C2 Cells Hypertrophy by Enhancing PPAR- Expression and Suppressing AP-1 Activity. PLoS ONE 2013, 8, e72548.
- Giaginis, C.; Tsourouflis, G.; Theocharis, S. Peroxisome Proliferator-Activated Receptor-gamma (PPAR-gamma) Ligands: Novel Pharmacological Agents in the Treatment of Ischemia Reperfusion Injury. Curr. Mol. Med. 2008, 8, 562–579.
- Liu, J.; Wang, L. Peroxisome proliferator-activated receptor gamma agonists for preventing recurrent stroke and other vascular events in people with stroke or transient ischaemic attack. Cochrane Database Syst. Rev. 2019, 2019.
- Liu, C.; Lee, T.; Lin, Y.; Sung, P.; Wei, Y.; Li, Y. Pioglitazone and PPAR-γ modulating treatment in hypertensive and type 2 diabetic patients after ischemic stroke: A national cohort study. Cardiovasc. Diabetol. 2020, 19, 2.
- Nesti, L.; Tricò, D.; Mengozzi, A.; Natali, A. Rethinking pioglitazone as a cardioprotective agent: A new perspective on an overlooked drug. Cardiovasc. Diabetol. 2021, 20, 109.
- Ren, Y.; Sun, C.; Sun, Y.; Tan, H.; Wu, Y.; Cui, B.; Wu, Z. PPAR gamma protects cardiomyocytes against oxidative stress and apoptosis via Bcl-2 upregulation. Vasc. Pharmacol. 2009, 51, 169–174.
- Satin, L.S.; Butler, P.C.; Ha, J.; Sherman, A.S. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol. Asp. Med. 2015, 42, 61–77.
- Rojas, J.; Chávez, M.; Olivar, L.; Rojas, M.; Morillo, J.; Mejías, J.; Calvo, M.; Bermúdez, V. Polycystic Ovary Syndrome, Insulin Resistance, and Obesity: Navigating the Pathophysiologic Labyrinth. Int. J. Reprod. Med. 2014, 2014, 719050.
- Sohn, K.K.; Cruciani-Guglielmacci, C.; Kassis, N.; Clément, L.; Ouali, F.; Caüzac, M.; Lebègue, N.; Berthelot, P.; Caignard, D.; Pégorier, J.; et al. S26948, a new specific peroxisome proliferator activated receptor gamma modulator improved in vivo hepatic insulin sensitivity in 48 h lipid infused rats. Eur. J. Pharmacol. 2009, 608, 104–111.
- Holman, R. Metformin as first choice in oral diabetes treatment: The UKPDS experience. J. Annu. Diabetol. Hotel. Dieu. 2007, 13–20.
- Harvey, I.; Stephens, J.M. Artemisia scoparia promotes adipogenesis in the absence of adipogenic effectors. Obesity 2021, 29, 1309–1319.
- Colca, J.R.; Scherer, P.E. The metabolic syndrome, thiazolidinediones, and implications for intersection of chronic and inflammatory disease. Mol. Metab. 2022, 55, 101409.
- Graham, D.J.; Green, L.; Senior, J.R.; Nourjah, P. Troglitazone-induced liver failure: A case study. Am. J. Med. 2003, 114, 299–306.
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. The Effect of Pioglitazone on Peroxisome Proliferator-Activated Receptor-gamma Target Genes Related to Lipid Storage In vivo. Diabetes Care 2004, 27, 1660–1667.
- Takahashi, Y.; Fukusato, T. Animal Models of Liver Diseases; Academic Press: Cambridge, MA, USA, 2017; pp. 313–339.
- Liu, C.; Feng, T.; Zhu, N.; Liu, P.; Han, X.; Chen, M.; Wang, X.; Li, N.; Li, Y.; Xu, Y.; et al. Identification of a novel selective agonist of PPARγ with no promotion of adipogenesis and less inhibition of osteoblastogenesis. Sci. Rep. 2015, 5, 9530.
- Pan, Y.; Zhao, D.; Yu, N.; An, T.; Miao, J.; Mo, F.; Gu, Y.; Zhang, D.; Gao, S.; Jiang, G. Curcumin improves glycolipid metabolism through regulating peroxisome proliferator activated receptor signalling pathway in high-fat diet-induced obese mice and 3T3-L1 adipocytes. R. Soc. Open Sci. 2017, 4, 170917.
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARγ and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510.
- Rőszer, T.; Menéndez-Gutiérrez, M.P.; Lefterova, M.I.; Alameda, D.; Núñez, V.; Lazar, M.A.; Fischer, T.; Ricote, M. Autoimmune Kidney Disease and Impaired Engulfment of Apoptotic Cells in Mice with Macrophage Peroxisome Proliferator-Activated Receptor gamma or Retinoid X Receptor Deficiency. J. Immunol. 2010, 186, 621–631.
- Hucke, S.; Floßdorf, J.; Grützke, B.; Dunay, I.R.; Frenzel, K.; Jungverdorben, J.; Linnartz, B.; Mack, M.; Peitz, M.; Brüstle, O.; et al. Licensing of myeloid cells promotes central nervous system autoimmunity and is controlled by peroxisome proliferator-activated receptor-γ. Brain 2012, 135, 1586–1605.
- Cheng, A.M.S.; Yin, H.Y.; Chen, A.; Liu, Y.; Chuang, M.; He, H.; Tighe, S.; Sheha, H.; Liao, S. Celecoxib and Pioglitazone as Potential Therapeutics for Regulating TGF-–Induced Hyaluronan in Dysthyroid Myopathy. Investig. Opthalmol. Vis. Sci. 2016, 57, 1951.
- Schmidt, S.; Moric, E.; Schmidt, M.; Sastre, M.; Feinstein, D.L.; Heneka, M.T. Anti-inflammatory and antiproliferative actions of PPAR- agonists on T lymphocytes derived from MS patients. J. Leukoc. Biol. 2003, 75, 478–485.
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J. Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPAR Agonist Treatment Approaches. Int. J. Mol. Sci. 2018, 19, 1212.
- Li, X.; Sun, Y.; Bao, J.; Chen, X.; Li, Y.; Yang, Y.; Zhang, L.; Huang, C.; Wu, B.; Meng, X.; et al. Functional role of PPAR-γ on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci. Rep. 2017, 7, 12671.
- Nozaki, Y.; Harada, K.; Sanzen, T.; Nakanuma, Y. PPAR ligand attenuates portal inflammation in the MRL-lpr mouse: A new strategy to restrain cholangiopathy in primary biliary cirrhosis. Med. Mol. Morphol. 2013, 46, 153–159.
- Bernardo, A.; Plumitallo, C.; Nuccio, C.D.; Visentin, S.; Minghetti, L. Curcumin promotes oligodendrocyte differentiation and their protection against TNF-α through the activation of the nuclear receptor PPAR-γ. Sci. Rep. 2021, 11, 4952.
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338.
- Khan, M.A.; Alam, Q.; Haque, A.; Ashafaq, M.; Khan, M.J.; Ashraf, G.M.; Ahmad, M. Current Progress on Peroxisome Proliferator-activated Receptor Gamma Agonist as an Emerging Therapeutic Approach for the Treatment of Alzheimer’s Disease: An Update. Curr. Neuropharmacol. 2019, 17, 232–246.
- Toobian, D.; Ghosh, P.; Katkar, G.D. Parsing the Role of PPARs in Macrophage Processes. Front. Immunol. 2021, 12, 783780.
- Salam, N.K.; Huang, T.H.; Kota, B.P.; Kim, M.S.; Li, Y.; Hibbs, D.E. Novel PPAR-gamma Agonists Identified from a Natural Product Library: A Virtual Screening, Induced-Fit Docking and Biological Assay Study. Chem. Biol. Drug Des. 2007, 71, 57–70.
- Xu, L.; Shen, S.; Ma, Y.; Kim, J.K.; Rodriguez-Agudo, D.; Heuman, D.M.; Hylemon, P.B.; Pandak, W.M.; Ren, S. 25-Hydroxycholesterol-3-sulfate attenuates inflammatory response via PPAR-γ signaling in human THP-1 macrophages. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E788–E799.
- Li, M.; Pascual, G.; Glass, C.K. Peroxisome Proliferator-Activated Receptor-γ Dependent Repression of the Inducible Nitric Oxide Synthase Gene. Mol. Cell. Biol. 2000, 20, 4699–4707.
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2007, 1771, 926–935.
- Petrova, T.V.; Akama, K.T.; Eldik, L.J.V. Cyclopentenone prostaglandins suppress activation of microglia: Down-regulation of inducible nitric-oxide synthase by 15-deoxy-Δ. Proc. Natl. Acad. Sci. USA 1999, 96, 4668–4673.
- Cuzzocrea, S.; Pisano, B.; Dugo, L.; Ianaro, A.; Maffia, P.; Patel, N.S.; Paola, R.D.; Ialenti, A.; Genovese, T.; Chatterjee, P.K.; et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-γ, reduces acute inflammation. Eur. J. Pharmacol. 2004, 483, 79–93.
- Wan, Y.; Evans, R.M. Rosiglitazone activation of PPARgamma suppresses fractalkine signaling. J. Mol. Endocrinol. 2009, 44, 135–142.
- Yessoufou, A.; Wahli, W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Med. Wkly. 2010, 140, w13071.
- Ramot, Y.; Mastrofrancesco, A.; Camera, E.; Desreumaux, P.; Paus, R.; Picardo, M. The role of PPARγ-mediated signalling in skin biology and pathology: New targets and opportunities for clinical dermatology. Exp. Dermatol. 2015, 24, 245–251.
- Dubrac, S.; Stoitzner, P.; Pirkebner, D.; Elentner, A.; Schoonjans, K.; Auwerx, J.; Sael, S.; Hengster, P.; Fritsch, P.; Romani, N.; et al. Peroxisome Proliferator-Activated Receptor-γ Activation Inhibits Langerhans Cell Function. J. Immunol. 2007, 178, 4362–4372.
- Sugiyama, H.; Nonaka, T.; Kishimoto, T.; Komoriya, K.; Tsuji, K.; Nakahata, T. Peroxisome proliferator-activated receptors are expressed in human cultured mast cells: A possible role of these receptors in negative regulation of mast cell activation. Eur. J. Immunol. 2000, 30, 3363–3370.
- Umeno, A.; Sakashita, M.; Sugino, S.; Murotomi, K.; Okuzawa, T.; Morita, N.; Tomii, K.; Tsuchiya, Y.; Yamasaki, K.; Horie, M.; et al. Comprehensive analysis of PPARγ agonist activities of stereo-, regio-, and enantio-isomers of hydroxyoctadecadienoic acids. Biosci. Rep. 2020, 40, BSR20193767.
- Emerson, M.R.; LeVine, S.M. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res. 2004, 1021, 140–145.
- Shankaranarayanan, P.; Nigam, S. IL-4 Induces Apoptosis in A549 Lung Adenocarcinoma Cells: Evidence for the Pivotal Role of 15-Hydroxyeicosatetraenoic Acid Binding to Activated Peroxisome Proliferator-Activated Receptor Transcription Factor. J. Immunol. 2003, 170, 887–894.
- Yamaguchi, A.; Tourdot, B.E.; Yeung, J.; Holman, T.; Holinstat, M.A. The 15-lipoxygenase-derived Oxylipins 15-HETrE And 15-HETE Inhibit Platelet Activation In Part Through Activation of PPARs. Arterioscler. Thromb. Vasc. Biol. 2021, 41, AP119.
- Li, Y.; Atkinson, K.; Zhang, T. Combination of chemotherapy and cancer stem cell targeting agents: Preclinical and clinical studies. Cancer Lett. 2017, 396, 103–109.
- Illés, P.; Grycová, A.; Krasulová, K.; Dvořák, Z. Effects of Flavored Nonalcoholic Beverages on Transcriptional Activities of Nuclear and Steroid Hormone Receptors: Proof of Concept for Novel Reporter Cell Line PAZ-PPARg. J. Agric. Food Chem. 2018, 66, 12066–12078.
- Mueller, C.; Weaver, V.; Heuvel, J.P.V.; August, A.; Cantorna, M.T. Peroxisome proliferator-activated receptor gamma ligands attenuate immunological symptoms of experimental allergic asthma. Arch. Biochem. Biophys. 2003, 418, 186–196.
- Liu, J.; Yao, Q.; Xie, X.; Cui, Q.; Jiang, T.; Zhao, Z.; Du, X.; Lai, B.; Xiao, L.; Wang, N. Procyanidin B2 Attenuates Nicotine-Induced Hepatocyte Pyroptosis through a PPAR-Dependent Mechanism. Nutrients 2022, 14, 1756.
- Choi, S.; Cha, B.; Iida, K.; Lee, Y.; Yonezawa, T.; Teruya, T.; Nagai, K.; Woo, J. Artepillin C, as a PPARγ ligand, enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Biochem. Pharmacol. 2011, 81, 925–933.
- Delebinski, C.I.; Twardziok, M.; Kleinsimon, S.; Hoff, F.; Mulsow, K.; Rolff, J.; Jäger, S.; Eggert, A.; Seifert, G. A Natural Combination Extract of Viscum album L. Containing Both Triterpene Acids and Lectins Is Highly Effective against AML In vivo. PLoS ONE 2015, 10, e0133892.
- Yang, L.; Zheng, Y.; Miao, Y.; Yan, W.; Geng, Y.; Dai, Y.; Wei, Z. Bergenin, a PPARγ agonist, inhibits Th17 differentiation and subsequent neutrophilic asthma by preventing GLS1-dependent glutaminolysis. Acta Pharmacol. Sin. 2021, 43, 963–976.
- Hong, J.; Samudio, I.; Chintharlapalli, S.; Safe, S. 1,1-bis(3′-indolyl)-1-(p-substituted phenyl)methanes decrease mitochondrial membrane potential and induce apoptosis in endometrial and other cancer cell lines. Mol. Carcinog. 2008, 47, 492–507.
- Yao, J.; Jiang, M.; Zhang, Y.; Liu, X.; Du, Q.; Feng, G. Chrysin alleviates allergic inflammation and airway remodeling in a murine model of chronic asthma. Int. Immunopharmacol. 2016, 32, 24–31.
- Wu, Q.; Needs, P.W.; Lu, Y.; Kroon, P.A.; Ren, D.; Yang, X. Different antitumor effects of quercetin, quercetin-3′-sulfate and quercetin-3-glucuronide in human breast cancer MCF-7 cells. Food Funct. 2018, 9, 1736–1746.
- Ballav, S.; Lokhande, K.B.; Dabhi, I.; Inje, S.; Ranjan, A.; Swamy, K.V.; Basu, S. Designing novel quercetin derivatives as matrix metalloproteinase-9 inhibitors in colon carcinoma: An In vitro and in silico approach. J. Dent. Res. Rev. 2020, 7, 30–35.
- Weng, J.; Bai, L.; Chiu, C.; Hu, J.; Chiu, S.; Wu, C. Cucurbitane Triterpenoid from Momordica charantia Induces Apoptosis and Autophagy in Breast Cancer Cells, in Part, through Peroxisome Proliferator-Activated Receptori-γ Activation. Evid.-Based Complement. Altern. Med. 2013, 2013, 935675.
- Noruddin, N.A.A.; Hamzah, M.F.; Rosman, Z.; Salin, N.H.; Shu-Chien, A.C.; Muhammad, T.S.T. Natural Compound 3,7,25-trihydroxycucurbita-5,23(E)-dien-19-al from Momordica charantia Acts as PPAR Ligand. Molecules 2021, 26, 2682.
- Kumar, R.; Balaji, S.; Uma, T.; Sehgal, P. Fruit extracts of Momordica charantia potentiate glucose uptake and up-regulate Glut-4, PPAR and PI3K. J. Ethnopharmacol. 2009, 126, 533–537.
- An, R.; Pathmanathan, K.; Shankernarayanan, N.; Vishwakarma, R.A.; Balakrishnan, A. Upregulation of Glut-4 and PPARgamma by an isoflavone from Pterocarpus marsupium on L6 myotubes: A possible mechanism of action. J. Ethnopharmacol. 2005, 97, 253–260.
- San, Y.; Liu, Y.; Zhang, Y.; Shi, P.; Zhu, Y. Peroxisome proliferator-activated receptor-γ agonist inhibits the mammalian target of rapamycin signaling pathway and has a protective effect in a rat model of status epilepticus. Mol. Med. Rep. 2015, 12, 1877–1883.
- Griggs, R.B.; Donahue, R.R.; Morgenweck, J.; Grace, P.M.; Sutton, A.; Watkins, L.R.; Taylor, B.K. Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPAR mechanisms. Pain 2015, 156, 469–482.
- Bongartz, T. Treatment of active psoriatic arthritis with the PPAR ligand pioglitazone: An open-label pilot study. Rheumatology 2005, 44, 126–129.
- Meng, X.; Sun, X.; Zhang, Y.; Shi, H.; Deng, W.; Liu, Y.; Wang, G.; Fang, P.; Yang, S. PPAR Agonist PGZ Attenuates OVA-Induced Airway Inflammation and Airway Remodeling via RGS4 Signaling in Mouse Model. Inflammation 2018, 41, 2079–2089.
- Khandoudi, N.; Delerive, P.; Berrebi-Bertrand, I.; Buckingham, R.E.; Staels, B.; Bril, A. Rosiglitazone, a Peroxisome Proliferator-Activated Receptor-, Inhibits the Jun NH2-Terminal Kinase/Activating Protein 1 Pathway and Protects the Heart From Ischemia/Reperfusion Injury. Diabetes 2002, 51, 1507–1514.
- Lee, K.S.; Park, S.J.; Hwang, P.H.; Yi, H.K.; Song, C.H.; Chai, O.H.; Kim, J.; Lee, M.K.; Lee, Y.C. PPAR-gamma modulates allergic inflammation through up-regulation of PTEN. FASEB J. 2005, 19, 1033–1035.
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Jin, S.M.; Lee, M.K.; Kim, U.H.; Lee, Y.C. Peroxisome proliferator activated receptor-γ modulates reactive oxygen species generation and activation of nuclear factor-B and hypoxia-inducible factor 1 in allergic airway disease of mice. J. Allergy Clin. Immunol. 2006, 118, 120–127.
- Wanguang, Z.; Huilan, Z.; Lihua, X. Influence of ciglitazone on A549 cells growth in vitro and in vivo and mechanism. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 36–39.
- Moss, P.E.; Lyles, B.E.; Stewart, L.V. The PPARγ ligand ciglitazone regulates androgen receptor activation differently in androgen-dependent versus androgen-independent human prostate cancer cells. Exp. Cell Res. 2010, 316, 3478–3488.
- Akinyeke, T.O.; Stewart, L.V. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPAR-γ independent mechanism. Cancer Biol. Ther. 2011, 11, 1046–1058.
- Ellis, C.N.; Varani, J.; Fisher, G.J.; Zeigler, M.E.; Pershadsingh, H.A.; Benson, S.C.; Chi, Y.; Kurtz, T.W. Troglitazone Improves Psoriasis and Normalizes Models of Proliferative Skin Disease. Arch. Dermatol. 2000, 136, 609–616.
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARγ. eLife 2018, 7, e43320.
- Thilakarathna, S.; Rupasinghe, H. Flavonoid Bioavailability and Attempts for Bioavailability Enhancement. Nutrients 2013, 5, 3367–3387.
- Horita, S.; Nakamura, M.; Satoh, N.; Suzuki, M.; Seki, G. Thiazolidinediones and Edema: Recent Advances in the Pathogenesis of Thiazolidinediones-Induced Renal Sodium Retention. PPAR Res. 2015, 2015, 646423.
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866.
- Filipova, E.; Uzunova, K.; Kalinov, K.; Vekov, T. Pioglitazone and the Risk of Bladder Cancer: A Meta-Analysis. Diabetes Ther. 2017, 8, 705–726.
- Tseng, C. Rosiglitazone may reduce thyroid cancer risk in patients with type 2 diabetes. Ann. Med. 2013, 45, 539–544.
- Tseng, C. Rosiglitazone reduces breast cancer risk in Taiwanese female patients with type 2 diabetes mellitus. Oncotarget 2016, 8, 3042–3048.
- Han, E.; Jang, S.; Kim, G.; Lee, Y.; Choe, E.Y.; Nam, C.M.; Kang, E.S. Rosiglitazone Use and the Risk of Bladder Cancer in Patients With Type 2 Diabetes. Medicine 2016, 95, e2786.
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411.
- Nissen, S.E.; Wolski, K. Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes. N. Engl. J. Med. 2007, 356, 2457–2471.
- Dormandy, J.; Bhattacharya, M.; van Troostenburg de Bruyn, A.R.; PROactive Investigators. Safety and Tolerability of Pioglitazone in High-Risk Patients with Type 2 Diabetes. Drug Saf. 2009, 32, 187–202.
- Nesto, R.W.; Bell, D.; Bonow, R.O.; Fonseca, V.; Grundy, S.M.; Horton, E.S.; Winter, M.L.; Porte, D.; Semenkovich, C.F.; Smith, S.; et al. Thiazolidinedione Use, Fluid Retention, and Congestive Heart Failure. Circulation 2003, 108, 2941–2948.
- Doshi, L.S.; Brahma, M.K.; Bahirat, U.A.; Dixit, A.V.; Nemmani, K.V. Discovery and development of selective PPAR gamma modulators as safe and effective antidiabetic agents. Expert Opin. Investig. Drugs 2010, 19, 489–512.
- Weidner, C.; de Groot, J.C.; Prasad, A.; Freiwald, A.; Quedenau, C.; Kliem, M.; Witzke, A.; Kodelja, V.; Han, C.; Giegold, S.; et al. Amorfrutins are potent antidiabetic dietary natural products. Proc. Natl. Acad. Sci. USA 2012, 109, 7257–7262.
- Aidhen, I.S.; Mukkamala, R.; Weidner, C.; Sauer, S. A Common Building Block for the Syntheses of Amorfrutin and Cajaninstilbene Acid Libraries toward Efficient Binding with Peroxisome Proliferator-Activated Receptors. Org. Lett. 2014, 17, 194–197.
- Ayza, M.A.; Zewdie, K.A.; Tesfaye, B.A.; Gebrekirstos, S.T.; Berhe, D.F. Anti-Diabetic Effect of Telmisartan Through its Partial PPAR-gamma Agonistic Activity. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3627–3635.
- Matsuyama, M.; Funao, K.; Kuratsukuri, K.; Tanaka, T.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.; Yoshimura, N.; Yoshimura, R. Telmisartan inhibits human urological cancer cell growth through early apoptosis. Exp. Ther. Med. 2010, 1, 301–306.
- Liu, Y.; Chen, S.; Liu, J.; Jin, Y.; Yu, S.; An, R. Telmisartan inhibits oxalate and calcium oxalate crystal-induced epithelial-mesenchymal transformation via PPAR-AKT/STAT3/p38 MAPK-Snail pathway. Life Sci. 2020, 241, 117108.
- Weidner, C.; Wowro, S.J.; Freiwald, A.; Kodelja, V.; Abdel-Aziz, H.; Kelber, O.; Sauer, S. Lemon balm extract causes potent antihyperglycemic and antihyperlipidemic effects in insulin-resistant obese mice. Mol. Nutr. Food Res. 2013, 58, 903–907.
- Zheng, W.; Feng, X.; Qiu, L.; Pan, Z.; Wang, R.; Lin, S.; Hou, D.; Jin, L.; Li, Y. Identification of the antibiotic ionomycin as an unexpected peroxisome proliferator-activated receptorγ (PPARγ) ligand with a unique binding mode and effective glucose-lowering activity in a mouse model of diabetes. Diabetologia 2012, 56, 401–411.
- Villacorta, L.; Schopfer, F.J.; Zhang, J.; Freeman, B.A.; Chen, Y.E. PPARγ and its ligands: Therapeutic implications in cardiovascular disease. Clin. Sci. 2009, 116, 205–218.
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing Effects of Retinoic Acid on Cell Growth Result from Alternate Activation of Two Different Nuclear Receptors. Cell 2007, 129, 723–733.
- Bray, G.A. The Zucker-fatty rat: A review. Fed. Proc. 1977, 36, 148–153.
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738.
- Coelho, M.S.; de Lima, C.L.; Royer, C.; Silva, J.B.; Oliveira, F.C.B.; Christ, C.G.; Pereira, S.A.; Bao, S.N.; Lima, M.C.A.; Pitta, M.G.R.; et al. GQ-16, a TZD-Derived Partial PPARgamma Agonist, Induces the Expression of Thermogenesis-Related Genes in Brown Fat and Visceral White Fat and Decreases Visceral Adiposity in Obese and Hyperglycemic Mice. PLoS ONE 2016, 11, e0154310.
- Ferreira, A.P.B.; Coelho, M.S.; Amato, A.A.; Neves, F.d.R.; Rodrigues, I.C.; Royer, C. Effect of PPAR Partial Agonist, GQ-16, on Viability of Breast Cancer Cells in Culture. FASEB J. 2017, 31, 876.5.
- Haraguchi, T.; Iwasaki, K.; Takasaki, K.; Uchida, K.; Naito, T.; Nogami, A.; Kubota, K.; Shindo, T.; Uchida, N.; Katsurabayashi, S.; et al. Telmisartan, a partial agonist of peroxisome proliferator-activated receptor gamma, improves impairment of spatial memory and hippocampal apoptosis in rats treated with repeated cerebral ischemia. Brain Res. 2010, 1353, 125–132.
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical role of PPARγ in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 2015, 7, 1529–1543.
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12.
- Phan, A.N.; Vo, V.T.; Hua, T.N.; Kim, M.; Jo, S.; Choi, J.; Kim, H.; Son, J.; Suh, Y.; Jeong, Y. PPAR sumoylation-mediated lipid accumulation in lung cancer. Oncotarget 2017, 8, 82491–82505.
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPAR Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174.
- Heudobler, D.; Rechenmacher, M.; Lüke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. Int. J. Mol. Sci. 2018, 19, 3540.
- Huang, C.; Lo, C.; Chiu, C.; Shyur, L. Deoxyelephantopin, a novel multifunctional agent, suppresses mammary tumor growth and lung metastasis and doubles survival time in mice. Br. J. Pharmacol. 2010, 159, 856–871.
- Gan, L.; Gan, Z.; Dan, Y.; Li, Y.; Zhang, P.; Chen, S.; Ye, Z.; Pan, T.; Wan, C.; Hu, X.; et al. Tetrazanbigen Derivatives as Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Partial Agonists:Design, Synthesis, Structure–Activity Relationship, and Anticancer Activities. J. Med. Chem. 2021, 64, 1018–1036.
- Gbelcová, H.; Švéda, M.; Laubertová, L.; Varga, I.; Vítek, L.; Kolář, M.; Strnad, H.; Zelenka, J.; Böhmer, D.; Ruml, T. The effect of simvastatin on lipid droplets accumulation in human embryonic kidney cells and pancreatic cancer cells. Lipids Health Dis. 2013, 12, 126.
- Joshi, H.; Marulkar, K.; Gota, V.; Ramaa, C.S. Hydroxy Cinnamic Acid Derivatives as Partial PPARγ Agonists: In silico Studies, Synthesis and Biological Characterization Against Chronic Myeloid Leukemia Cell Line (K562). Anti-Cancer Agents Med. Chem. 2017, 17, 524–541.
- Kim, M.; Chae, Y.N.; Choi, S.; Moon, H.S.; Son, M.; Bae, M.; Choi, H.; Hur, Y.; Kim, E.; Park, Y.H.; et al. PAM-1616, a selective peroxisome proliferator-activated receptor modulator with preserved anti-diabetic efficacy and reduced adverse effects. Eur. J. Pharmacol. 2011, 650, 673–681.
- Fujimura, T.; Kimura, C.; Oe, T.; Takata, Y.; Sakuma, H.; Aramori, I.; Mutoh, S. A Selective Peroxisome Proliferator-Activated Receptor Modulator with Distinct Fat Cell Regulation Properties. J. Pharmacol. Exp. Ther. 2006, 318, 863–871.
- Konda, V.R.; Desai, A.; Darland, G.; Grayson, N.; Bland, J.S. KDT501, a Derivative from Hops, Normalizes Glucose Metabolism and Body Weight in Rodent Models of Diabetes. PLoS ONE 2014, 9, e87848.
- Smith, A.G.; Beaumont, K.A.; Smit, D.J.; Thurber, A.E.; Cook, A.L.; Boyle, G.M.; Parsons, P.G.; Sturm, R.A.; Muscat, G.E. PPAR agonists attenuate proliferation and modulate Wnt/-catenin signalling in melanoma cells. Int. J. Biochem. Cell Biol. 2009, 41, 844–852.
- Li, J.; Chen, L.; Yu, P.; Liu, B.; Zhu, J.; Yang, Y. Telmisartan Exerts Anti-Tumor Effects by Activating Peroxisome Proliferator-Activated Receptor- in Human Lung Adenocarcinoma A549 Cells. Molecules 2014, 19, 2862–2876.
- Higgins, L.S.; Mantzoros, C.S. The Development of INT131 as a Selective PPARgamma Modulator: Approach to a Safer Insulin Sensitizer. PPAR Res. 2008, 2008, 936906.
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγPartial Agonism. PPAR Res. 2015, 2015, 816856.
- Laghezza, A.; Montanari, R.; Lavecchia, A.; Piemontese, L.; Pochetti, G.; Iacobazzi, V.; Infantino, V.; Capelli, D.; DeBellis, M.; Liantonio, A.; et al. On the Metabolically Active Form of Metaglidasen: Improved Synthesis and Investigation of Its Peculiar Activity on Peroxisome Proliferator-Activated Receptors and Skeletal Muscles. ChemMedChem 2015, 10, 555–565.
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189.
- Chen, L.; Bush, C.R.; Necela, B.M.; Su, W.; Yanagisawa, M.; Anastasiadis, P.Z.; Fields, A.P.; Thompson, E.A. RS5444, a novel PPARgamma agonist, regulates aspects of the differentiated phenotype in nontransformed intestinal epithelial cells. Mol. Cell. Endocrinol. 2006, 251, 17–32.
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413.
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an Oral PPAR-gamma Agonist, in Combination With Paclitaxel in Anaplastic Thyroid Cancer: Results of a Multicenter Phase 1 Trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400.

