 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roberta Misasi | + 2348 word(s) | 2348 | 2020-11-17 08:39:29 | | | |
| 2 | Vivi Li | -52 word(s) | 2296 | 2020-11-23 10:43:27 | | |
Video Upload Options
Antiphospholipid Syndrome (APS) is an autoimmune disease characterized by arterial and/or venous thrombosis and/or pregnancy morbidity, associated with circulating antiphospholipid antibodies (aPL).
1. Introduction
APS is an autoimmune disease, characterized by recurrent thrombosis and/or obstetrical morbidity and a series of systemic manifestations induced by the persistent presence of aPL, including lupus anticoagulant (LA), anti-β2-GPI and/or anti-cardiolipin (aCL) antibodies.
Classification criteria for diagnosis of APS require that a patient has the combination of at least one laboratory and one clinical (arterial or venous thrombosis and/or pregnancy morbidity) criteria, as reported by the revised international classification criteria for APS [1][2][3]. However, the clinical spectrum of the disease may include additional manifestations which may affect various organs and cannot be explained exclusively by a prothrombotic state [4].
APS often occurs as a secondary disease in association with other autoimmune disorders, mostly systemic lupus erythematosus (SLE), in these cases it is usually referred to as “secondary” APS (SAPS), to be distinguished from the “primary” form called PAPS.
In the last decade, the significance of aPL persistency and accumulation (the co-presence of aPL criteria: aCL, anti-β2-GPI of the IgG or IgM subtypes and circulating LAC) was evaluated, especially regarding the risk of APS evolvement, although their functional role for assessment of the specific APS-related manifestations is not always clear. However, “seropositivity” of all the three-classification criteria, termed the “triple positive”-variant, correlates with a more aggressive disease. It requires specific therapeutic interventions, such as anti-coagulant drugs [5][6]. Several studies revealed that aPL are a heterogeneous group of autoantibodies that have a clear association with thrombosis and pregnancy morbidity and are directed against proteins, complexes of phospholipids and phospholipid-binding proteins or phospholipids [7][8]. Proteins as a target of “aPL antibodies” are so far identified not only as β2-GPI [9], but also as annexin A5 [10], annexin A2 [11], prothrombin (PT) [12], protein C [13] and protein S [14]; as complexes of phospholipids and phospholipid-binding proteins, prothrombin/phosphatidylserine (PT/PS) [15] and vimentin/cardiolipin [16]; as phospholipids, in addition to cardiolipin, phosphatidylserine (PS) [7] phosphatidylethanolamine (PE) [17] and lysobisphosphatidic acid (LBPA) [18].
However, in daily clinical practice, it is possible to identify patients with clinical symptoms suggestive for APS, but persistently negative for classical laboratory criteria. This patients’ population was referred to as seronegative SN-APS, although “new” aPL specificities have been described in these patients. In fact, in 2003 it was deemed necessary to classify a new entity of the syndrome (SN-APS) that it is still the subject of debate. G. Hughes, M. Khamashta and other research teams speculated that this clinical entity could be explained by the presence of non-criteria aPL, which may not have been considered in the serological battery of the tests [19][20].
Despite the strong association between aPL and thrombosis, the exact pathogenic mechanisms underlying thrombotic events and pregnancy morbidity in the course of APS have not been yet fully elucidated and more than one mechanism may be involved, such as the exposure to some environmental agents, such as infections, in susceptible individuals [21][22][23]
Antiphospholipid antibodies binding β2-GPI, may contribute to thrombotic diathesis by interfering with hemostasis [24][25]. Numerous studies highlight the role of activation of monocytes, endothelial cells, platelets and/or complement, as well as the induction of a prothrombotic state caused by interference with coagulation cascade proteins. aPL interact with endothelial cells inducing adhesion molecules, such as intercellular cell adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin expression and proinflammatory cytokines release, thus inducing a proinflammatory and procoagulant cell phenotype [22] Moreover, aPL may activate platelets, with an increase of glycoprotein IIb–IIIa expression [23], thromboxane A2 synthesis [26] and platelet factor-4 secretion, a chemokine with procoagulant and prothrombotic effects [27][28]. Several reports show that in monocytes and endothelial cells, anti-β2-GPI antibodies may lead to an up-regulation of Tissue Factor (TF), which plays a pivotal role in initiating the extrinsic coagulation cascade [29].
Another mechanism involves the Annexin A5 protein, this is a natural and physiological anti-coagulant which binds to PS on the cell surface, and forms a shield to prevent the activation of procoagulant complexes. Anti-β2-GPI/β2-GPI complex can disrupt this anticoagulant shield, exposing procoagulant PS, hence predisposing to thrombosis [30][31].
To explain the pathogenesis of thrombosis in APS, a model was suggested: “the first hit and second hit”. According to this model, aPL (the “first hit”) destroy the integrity of the endothelium inducing a procoagulant phenotype, nevertheless, thrombosis takes place only in the presence of an initiating factor (the “second hit”), as a consequence of smoking, infection, oxidative stress or inflammation [32]. Thus, in some cases, aPL cannot be considered pathogenic in those patients who are termed as “asymptomatic carriers” [33]. One of the molecular mechanism involving endothelial cells and compatible with this pathogenetic hypothesis is represented by the activation and signaling through Toll-like Receptor 4 (TLR-4) that drives ultimately to an excessive release of proinflammatory cytokines (“cytokine storm”) and an increase in the production of procoagulant factors, as well as the expression of cell-adhesion molecules [34][35].
2. β2-GPI Conformations in the Pathogenesis of APS
The pathogenetic mechanism of aPL and mostly anti-β2-GPI antibodies includes, among other mechanisms, the alteration of both phases of the coagulation cascade: the fluid phase, by interfering with the Vitamin K dependent protein C and annexin A5, and the cellular phase, by activating platelets, inducing the expression of TF on monocytes and endothelial cells with consequent activation of complement [4][24].
β2-GPI, also known as apolipoprotein H, is a 45 kDa plasma glycoprotein with affinity for anionic phospholipids. It is mainly synthetized in the liver and has a plasma concentration of approximately 200 μg/mL. It has been suggested that β2-GPI may inhibit platelet prothrombinase activity [36] and ADP-mediated platelet aggregation [37]. Moreover, because of its high affinity for anionic phospholipids, it was thought that β2-GPI, by inhibition of the contact phase activation of coagulation, could play a role in maintaining the hemostatic balance [38]. It has also been suggested that β2-GPI may function as a scavenger protein. Indeed, the molecule was shown to react with lipopolysaccharide (LPS), facilitating its phagocytosis by monocytes-macrophages [39]. Consistent with such scavenging activity is also the binding of β2-GPI to apoptotic material. During apoptosis, the reorganization of membrane lipid bilayer determines the exposure of PS, which is generally distributed in the cytoplasmic leaflet of the plasma membrane of cells, on the outer cell surface. β2-GPI binds to PS-exposing vesicles or apoptotic cells, promoting their engulfment by phagocytes [40].
Although β2-GPI was first described in 1961 by Schultze et al. [41], the interest in this protein increased significantly only about thirty years after its discovery, when it was recognized as the major antigenic target in APS [42][43][44]. β2-GPI consists of a single polypeptide chain of 326 amino acid residues and comprises five domains, arranged like a pearl necklace. The first four domains are short consensus repeats from the complement control protein (CCP) module type, also known as “sushi domains”. CCPs are modules of about 60 amino acids common in many proteins involved in the regulation of complement activation. On the contrary, the amino acid sequence of the fifth domain (DV) gives it features different from the other domains. The DV, in fact, is stabilized by three internal disulfide bonds instead of two. It carries a definite positive charge and two portions, located at the lower part of the DV, constitute an excellent counterpart for interactions with negatively charged amphiphilic substances. Therefore, the DV is responsible for the binding of β2-GPI to anionic phospholipids on cell membranes. β2-GPI exists in two interconvertible biochemical variants, oxidized (54%) and reduced (46%), depending on the integrity of the disulfide bonds [45][46]. In the oxidized form, 11 disulfide bonds are formed. In the reduced form, the disulfide bonds C288–C326 in DV and C32–C60 in DI are individually or simultaneously broken. After the acknowledgment of the central importance of β2-GPI in APS, many studies followed to identify the portions of the protein involved in the binding of anti-β2-GPI antibodies. It has been shown that antibodies can be directed against epitopes located in different domains of the protein. However, robust evidence indicates that the immunodominant epitope is in the first domain (DI) of the protein. Using domain-deletion mutants, Iverson et al. showed for the first time that anti-β2-GPI antibodies recognize an epitope on DI [47]. To further explore the fine specificity of these anti-DI antibodies, Iverson et al. constructed several other mutants with point mutations in domain I [48]. The main epitope was reported to involve the amino acid residues R39-R43 and D8-D9, as well as the linker region between domains I and II.
Multiple studies have led to the identification of anti-β2-GPI DI antibodies as the key pathogenic subset of autoantibodies in APS. In vitro, anti-β2-GPI DI IgG were found to have lupus LAC activity and strongly associate with thrombosis. Moreover, recombinant human β2-GPI DI was found to lower aPL-induced thrombosis [49]. Furthermore, anti-β2-GPI DI IgG have been shown to confer increased resistance to the anticoagulant properties of both annexin A5 [50] and activated protein C [51]. Anti-β2-GPI antibodies displaying LAC activity were also demonstrated to abrogate the β2-GPI-mediated inhibitory effect on von Willebrand factor dependent platelet adhesion and aggregation [52]. On the other hand, they may also interfere with the intrinsic anti-thrombotic functions of β2-GPI. Importantly, both affinity-purified anti-β2GPI DI IgG from APS serum and human monoclonal anti-β2GPI DI IgG have been shown to induce thrombosis and/or fetal loss in mice APS models of venous thrombosis [53][54]. Agostinis et al. proved that a human monoclonal antibody directed against β2-GPI DI exhibits complement-dependent procoagulant and pro-abortive effects, and a variant of this antibody, lacking the CH2 domain, is effective in preventing blood clot formation and fetal loss induced by aPL [55]. Therefore, it is now widely accepted that autoantibodies directed against β2-GPI DI can drive APS pathogenesis and are associated with thromboembolic events. In a large cohort study, Andreoli et al. have demonstrated that IgG targeting β2-GPI DI represent the prevalent subset not only among APS patients but also among individuals with autoimmune conditions with any clinical sign indicative of APS. They proposed that the ratio between antibodies to DI and those targeting DIV/V of β2-GPI may be useful in determining the pathogenic potential of anti-β2-GPI antibodies and in discriminating between autoimmune disorders and non-immune conditions [56]. Although it is widely recognized that the first domain of β2-GPI plays a fundamental role in the pathogenesis of the syndrome, multiple studies suggest that aPL directed against different domains of the protein may play a pathogenic role [57]. In particular, Murthy et al. demonstrated that IgA anti-β2-GPI directed to domain IV/V of the molecule represent an important subgroup of clinically relevant aPL, which may play a pathogenic role, as revealed in a mouse model of thrombosis [58]. Other researchers have recently shown that IgA anti-β2-GPI antibodies found in APS patients with clinical signs of thrombosis bind to three sites in D3, D4 and D5 [59]. Moreover, anti-β2GPI (ILA-1, ILA-3 and H-3 MoAb), that are able to interact specifically with three different hexapeptides corresponding to distinct epitopes located in domains I-II, III and IV of the molecule [60][61], are able to activate endothelial cells in vitro and induce experimental APS by passive transfer [62].
The antigenic epitopes of β2-GPI domains have been defined as cryptic, since they are exposed on the outer surface of the protein only when the latter is in the open conformation. On the contrary, these epitopes are either buried by domain V in the circular form or shielded by the N-linked glycosylations in the S-twisted form. Indeed, β2-GPI can adopt multiple conformations (J-elongated, S-twisted and O-circular), resulting in different exposures of each of its domains to the solvent [63][64]. The crystal structure of β2-GPI revealed a hockey stick-like shape of the molecule, in which the first four domains are stretched along their long axis while the fifth domain is at a right angle to the other ones. This structure is thought to be the immunogenic conformation of β2-GPI that interacts with anti-β2-GPI antibodies, which forms when the protein binds to the membranes. The O-circular structure of β2-GPI was originally proposed by Koike et al. in 1998 [65] to explain the lack of binding of antibodies to β2-GPI in solution, and then it was captured by electron microscopy by Agar et al. [63]. This closed circular conformation has been shown to result from an intramolecular interaction between DI and DV, which in turn causes the epitope on DI to be hidden by DV. It is believed to be the conformation that the protein takes in the free form in plasma.
All these structural studies have led to the conclusion that in vivo β2-GPI can exist in both closed and open conformations [63], and that the interaction of the molecule with the surrounding microenvironment determines its structure at any given time. Specifically, the commonly accepted model predicts that in plasma β2-GPI is present primarily (>90%) in the closed circular conformation, with the epitope on domain I not accessible for autoantibodies. This assumption is consistent with the observation that circulating immunocomplexes between β2-GPI and antibodies are usually not easily detected in APS patients’ sera. In contrast, when β2-GPI interacts with negatively charged surfaces, such as anionic phospholipids, a conformational change occurs and β2-GPI adopts an open elongated form; the epitope on domain I is exposed to the solvent and antibodies can recognize it and bind to β2-GPI (Figure 1).
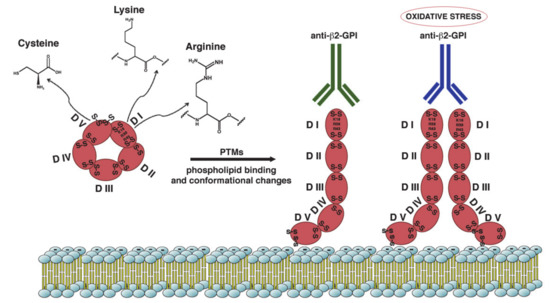
Figure 1. Schematic representation of β2-GPI structure and conformation. β2-GPI consist in five domains (I-V) with two disulfide bonds in each domain and an additional disulfide bond in domain V. Phospholipid binding by domain V and some PTMs of the protein result in a conformational change from the circular (closed) form to open configuration. This unfolded conformation may facilitate the exposition of ‘‘cryptic epitope’’ and autoantibodies binding. The amino acids most involved in the PTMs are Lysine, Arginine and Cysteine. Increased oxidative stress may alter the configuration of β2-GPI to a dimeric form that enhance antibody affinity.
References
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; Groot, P.G.D.E.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306.
- Cervera, R.; Piette, J.C.; Font, J.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; Lakos, G.; Tincani, A.; Kontopoulou-Griva, I.; et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1000 patients. Arthritis Rheum. 2002, 46, 1019–1027.
- Ruiz-Irastorza, G.; Crowther, M.; Branch, W.; Khamashta, M.A. Antiphospholipid syndrome. Lancet 2010, 376, 1498–1509.
- Negrini, S.; Pappalardo, F.; Murdaca, G.; Indiveri, F.; Puppo, F. The antiphospholipid syndrome: From pathophysiology to treatment. Clin. Exp. Med. 2017, 17, 257–267.
- Yelnik, C.M.; Urbanski, G.; Drumez, E.; Sobanski, V.; Maillard, H.; Lanteri, A.; Morell-Dubois, S.; Caron, C.; Dubucquoi, S.; Launay, D.; et al. Persistent triple antiphospho lipid antibody positivity as a strong risk factor of frst thrombosis, in a long-term follow-up study of patients without history of thrombosis or obstetrical morbidity. Lupus 2017, 26, 163–169.
- Otomo, K.; Atsumi, T.; Amengual, O.; Fujieda, Y.; Kato, M.; Oku, K.; Horita, T.; Yasuda, S.; Koike, T. Efficacy of the antiphospholipid score for the diagnosis of antiphospholipid syndrome and its predictive value for thrombotic events. Arthritis Rheum. 2012, 64, 504–512.
- Favaloro, E.J.; Wong, R.C.W. Antiphospholipid antibody testing for the antiphospholipid syndrome: A comprehensive practical review including a synopsis of challenges and recent guidelines. Pathology 2014, 46, 481–495.
- Misasi, R.; Capozzi, A.; Longo, A.; Recalchi, S.; Lococo, E.; Alessandri, C.; Conti, F.; Valesini, G.; Sorice, M. “New” Antigenic Targets and Methodological Approaches for Refining Laboratory Diagnosis of Antiphospholipid Syndrome. J. Immunol. Res. 2015, 2015, 858542.
- De Laat, H.B.; Derksen, R.H.W.M.; De Groot, P.G. beta2-glycoprotein I, the playmaker of the antiphospholipid syndrome. Clin. Immunol. 2004, 112, 161–168.
- Kaburaki, J.; Kuwana, M.; Yamamoto, M.; Kawai, S.; Ikeda, Y. Clinical significance of anti-annexin V antibodies in patients with systemic lupus erythematosus. Am. J. Hematol. 1997, 54, 209–213.
- Salle, V.; Mazie`re, J.C.; Smail, A.; Cévallos, R.; Mazière, C.; Fuentes, V.; Tramier, B.; Makdassi, R.; Choukroun, G.; Vittecoq, O.; et al. Anti-annexin II antibodies in systemic autoimmune diseases and antiphospholipid syndrome. J. Clin. Immunol. 2008, 28, 291–297.
- Arvieux, J.; Darnige, L.; Caron, C.; Reber, G.; Bensa, J.C.; Colomb, M.G. Development of an ELISA for autoantibodies to prothrombin showing their prevalence in patients with lupus anticoagulants. Thromb. Haemost. 1995, 74, 1120–1125.
- Oosting, J.D.; Derksen, R.H.W.M.; Bobbink, I.W.G.; Hackeng, T.M.; Bouma, B.N.; De Groot, P.G. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: An explanation for their pathogenic mechanism? Blood 1993, 81, 2618–2625.
- Sorice, M.; Arcieri, P.; Griggi, T.; Circella, A.; Misasi, R.; Lenti, L.; Di Nucci, G.D.; Mariani, G. Inhibition of protein S by autoantibodies in patients with acquired protein S deficiency. Thromb. Haemost. 1996, 75, 555–559.
- Sciascia, S.; Sanna, G.; Murru, V.; Roccatello, D.; Khamashta, M.A.; Bertolaccini, M.L. Anti-prothrombin (aPT) and anti- phosphatidylserine/prothrombin (aPS/PT) antibodies and the risk of thrombosis in the antiphospholipid syndrome. A sys- tematic review. Thromb. Haemost. 2013, 111, 354–364.
- Ortona, E.; Capozzi, A.; Colasanti, T.; Conti, F.; Alessandri, C.; Longo, A.; Garofalo, T.; Margutti, P.; Misasi, R.; Khamashta, M.A.; et al. Vimentin/cardiolipin complex as a new antigenic target of the antiphospholipid syndrome. Blood 2010, 116, 2960–2967.
- Sanmarco, M.; Gayet, S.; Alessi, M.C.; Audrain, M.; de Maistre, E.; Gris, J.C.; de Groot, P.G.; Hachulla, E.; Harlé, J.R.; Sié, P.; et al. Antiphosphatidylethanolamine antibodies are associated with an increased odds ratio for thrombosis—A multicenter study with the partici- pation of the European Forum on antiphospholipid antibodies. Thromb. Haemost. 2007, 97, 949–954.
- Alessandri, C.; Bombardieri, M.; Di Prospero, L.; Conigliaro, P.; Conti, F.; Labbadia, G.; Misasi, R.; Sorice, M.; Valesini, G. Anti-lysobisphosphatidic acid antibodies in patients with antiphospholipid syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2005, 140, 173–180.
- Hughes, G.R.V.; Khamashta, M.A. Seronegative antiphospholipid syndrome. Ann. Rheum. Dis. 2003, 62, 1127.
- Sciascia, S.; Amigo, M.; Roccatello, D.; Khamashta, M.A. Diagnosing antiphospholipid syndrome:’extra-criteria’manifestations and technical advances. Nat. Rev. Rheumatol. 2017, 13, 548–560.
- Galli, M.; Barbui, T.; Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Antiphospholipid antibodies: Involvement of protein cofactors. Haematologica 1993, 78, 1–4.
- Pierangeli, S.S.; Chen, P.P.; Raschi, E.; Scurati, S.; Grossi, C.; Borghi, M.O.; Palomo, I.; Harris, E.N.; Meroni, P.L. Antiphospholipid antibodies and the antiphospholipid syndrome: Pathogenic mechanisms. Semin. Thromb. Hemost. 2008, 34, 236–250.
- Jimenez, S.; Tassies, D.; Espinosa, G.; García-Criado, A.; Plaza, J.; Monteagudo, J.; Cervera, R.; Reverter, J.C. Double heterozygosity polymorphisms for platelet glycoproteins Ia/IIa and IIb/IIIa increases arterial thrombosis and arteriosclerosis in patients with the antiphospholipid syndrome or with systemic lupus erythematosus. Ann. Rheum. Dis. 2008, 67, 835–840.
- Arachchillage, D.R.J.; Laffan, M. Pathogenesis and management of antiphospholipid syndrome. Br. J. Haematol. 2017, 178, 181–195.
- Galli, M.; Comfurius, P.; Maassen, C.; Hemker, H.C.; De Baets, M.H.; Van Breda-Vriesman, P.J.; Barbui, T.; Zwaal, R.F.; Bevers, E.M. Anticardiolipin anti- bodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990, 335, 1544–1547.
- Robbins, D.L.; Leung, S.; Miller-Blair, D.J.; Ziboh, V. Effect of anticardiolipin/beta2-glycoprotein I complexes on production of thromboxane A2 by platelets from patients with the antiphospholipid syndrome. J. Rheumatol. 1998, 25, 51–56.
- Sikara, M.P.; Routsias, J.G.; Samiotaki, M.; Panayotou, G.; Moutsopoulos, H.M.; Vlachoyiannopoulos, P.G. {beta}2 Glycoprotein I ({beta}2GPI) binds platelet factor 4 (PF4): Implications for the pathogenesis of antiphospholipid syndrome. Blood 2010, 115, 713–723.
- Zucker, M.B.; Katz, I.R. Platelet factor 4: Production, structure, and physiologic and immunologic action. Proc. Soc. Exp. Biol. Med. 1991, 198, 693–702.
- Amengual, O.; Atsumi, T.; Khamashta, M.A.; Hughes, G.R. The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb. Haemost. 1998, 79, 276–281.
- Rand, J.H.; Wu, X.X.; Andree, H.A.; Lockwood, C.J.; Guller, S.; Scher, J.; Harpel, P.C. Pregnancy loss in the antiphospholipid-antibody syndrome—a possible thrombogenic mechanism. N. Engl. J. Med. 1997, 337, 154–160.
- Rand, J.H.; Wu, X.X.; Quinn, A.S.; Ashton, A.W.; Chen, P.P.; Hathcock, J.J.; Andree, H.A.M.; Taatjes, D.J. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: Evidence for a novel effect for an old antimalarial drug. Blood 2010, 115, 2292–2299.
- Meroni, P.; Borghi, M.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339.
- Linnemann, B. Antiphospholipid Syndrome-an update. Vasa 2018, 47, 451–464.
- Meroni, P.L.; Raschi, E.; Camera, M.; Testoni, C.; Nicoletti, F.; Tincani, A.; Khamashta, M.A.; Balestrieri, G.; Tremoli, E.; Hess, D.C. Endothelial activation by aPL: A potential pathogenetic mechanism for the clinical manifestations of the syndrome. J. Autoimmun. 2000, 15, 237–240.
- Raschi, E.; Testoni, C.; Bosisio, D.; Borghi, M.O.; Koike, T.; Mantovani, A.; Meroni, P.L. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood 2003, 101, 3495–3500.
- Nimpf, J.; Bevers, E.M.; Bomans, P.H.; Till, U.; Wurm, H.; Kostner, G.M.; Zwaal, R.F. Prothrombinase activity of human platelets is inhibited by beta 2-glycoprotein-I. Biochim. Biophys. Acta 1986, 884, 142–149.
- Nimpf, J.; Wurm, H.; Kostner, G.M. Interaction of b2-glycoprotein-I with human blood platelets: Influence upon the ADP-induced aggregation. Thromb. Haemost. 1985, 54, 397–401.
- Shi, T.; Giannakopoulos, B.; Iverson, G.M.; Cockerill, K.A.; Linnik, M.D.; Krilis, S.A. Domain V of beta2-glycoprotein I binds factor XI/XIa and is cleaved at Lys317-Thr318. J. Biol. Chem. 2005, 280, 907–912.
- Agar, C.; de Groot, P.G.; Mo¨rgelin, M.; Monk, S.D.D.C.; van Os, G.M.A.; Levels, J.H.M.; de Laat, B.; Urbanus, R.T.; Herwald, H.; van der Poll, T.; et al. β2-glycoprotein I: A novel component of innate immunity. Blood 2011, 117, 6939–6947.
- Balasubramanian, K.; Schroit, A.J. Characterization of phosphatidylserine-dependent beta2-glycoprotein I macrophage interactions. Implications for apoptotic cell clearance by phagocytes. J. Biol. Chem. 1998, 273, 29272–29277.
- Schultze, H.E.; Heide, K.; Haupt, H. Uber ein bischer unb ekanntesniedermolekularis b2-globulins des human serums. Naturwissens-chaften 1961, 48, 719.
- De Groot, P.G.; Meijers, J. β(2) -Glycoprotein I: Evolution, structure and function. J. Thromb. Haemost. 2011, 9, 1275–1284.
- McNeil, H.P.; Simpson, R.J.; Chesterman, C.N.; Krilis, S.A. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: Beta 2-glycoprotein I (apolipoprotein H). Proc. Natl. Acad. Sci. USA 1990, 87, 4120–4124.
- Matsuura, E.; Igarashi, Y.; Fujimoto, M.; Ichikawa, K.; Koike, T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet 1990, 336, 177–178.
- Ioannou, Y.; Zhang, J.Y.; Qi, M.; Gao, L.; Qi, J.C.; Yu, D.M.; Lau, H.; Sturgess, A.D.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M.; et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen β2-glycoprotein I. Arthritis Rheum. 2011, 63, 2774–2782.
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 2013, 368, 1033–1044.
- Iverson, G.M.; Victoria, E.J.; Marquis, D.M. Anti-beta2 glycoprotein I(beta2GPI) autoantibodies recognize an epitope on the first domain of beta2GPI. Proc. Natl. Acad. Sci. USA 1998, 95, 15542–15546.
- Iverson, G.M.; Reddel, S.; Victoria, E.J.; Cockerill, K.A.; Wang, Y.X.; Marti-Renom, M.A.; Sali, A.; Marquis, D.M.; Krilis, S.A.; Linnik, M.D. Use of single point mutations in domain I of beta 2-glycoprotein I to determine fine antigenic specificity of antiphospholipid autoantibodies. J. Immunol. 2002, 169, 7097–7103.
- Ioannou, Y.; Romay-Penabad, Z.; Pericleous, C.; Giles, I.; Papalardo, E.; Vargas, G.; Shilagard, T.; Latchman, D.S.; Isenberg, D.A.; Rahman, A. In vivo inhibition of antiphospholipid antibody induced pathogenicity utilizing the antigenic target peptide domain I of beta2-glycoprotein I: Proof of concept. J. Thromb. Haemost. 2009, 7, 833–842.
- De Laat, B.; Wu, X.X.; Van Lummel, M.; Derksen, R.H.W.M.; De Groot, P.G.; Rand, J.H. Correlation between antiphospholipid antibodies that recognize domain I of β2-glycoprotein I and a reduction in the anticoagulant activity of annexin A5. Blood 2007, 109, 1490–1494.
- Gardiner, C.; Cohen, H.; Jenkins, A.; Machin, S.J.; Mackie, I.J. Detection of acquired resistance to activated protein C associated with antiphospholipid antibodies using a novel clotting assay. Blood Coagul. Fibrinolysis 2006, 17, 477–478.
- Hulstein, J.J.; Lenting, P.J.; De Laat, B.; Derksen, R.H.; Fijnheer, R.; De Groot, P.G. beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood 2007, 110, 1483–1491.
- Pierangeli, S.S.; Liu, X.; Espinola, R.; Zhu, M.; Harris, N.E.; Chen, P.P. Functional analyses of patient-derived IgG monoclonal anticardiolipin antibodies using in vivo thrombosis and in vivo microcirculation models. Thromb. Haemost. 2000, 84, 388–395.
- Giles, I.; Pericleous, C.; Liu, X.; Ehsanullah, J.; Clarke, L.; Brogan, P.; Newton-West, M.; Swerlick, R.; Lambrianides, A.; Chen, P. Thrombin binding predicts the effects of sequence changes in a human monoclonal antiphospholipid antibody on its in vivo biologic actions. J. Immunol. 2009, 182, 4836–4843.
- Agostinis, C.; Durigutto, P.; Sblattero, D.; Borghi, M.O.; Grossi, C.; Guida, F.; Bulla, R.; Macor, P.; Pregnolato, F.; Meroni, P.L.; et al. A non complement-fixing antibody to beta2 glycoprotein I as a novel therapy to control abortions and thrombosis in antiphospholipid syndrome. Blood 2014, 123, 3478–3487.
- Andreoli, L.; Nalli, C.; Motta, M.; Norman, G.L.; Shums, Z.; Encabo, S.; Binder, W.L.; Nuzzo, M.; Frassi, M.; Lojacono, A. Anti-b2-glycoprotein I IgG antibodies from 1-year-old healthy children born to mothers with systemic autoimmune diseases preferentially target domain 4/5: Might it be the reason for their ‘innocent’ profile? Ann. Rheum. Dis. 2011, 70, 380–383.
- Giles, I.P.; Isenberg, D.A.; Latchman, D.S.; Rahman, A. How do antiphospholipid antibodies bind beta2-glycoprotein I? Arthritis Rheum. 2003, 48, 2111–2121.
- Murthy, V.; Willis, R.; Romay-Penabad, Z.; Ruiz-Limón, P.; Martínez-Martínez, L.A.; Jatwani, S.; Jajoria, P.; Seif, A.; Alarcón, G.S.; Papalardo, E.; et al. Value of isolated IgA anti-β2 -glycoprotein I positivity in the diagnosis of the antiphospholipid syndrome. Arthritis Rheum. 2013, 65, 3186–3193.
- Serrano, M.; Martinez-Flores, J.A.; Norman, G.L.; Naranjo, L.; Morales, J.M.; Serrano, A. The IgA Isotype of Anti-β2 Glycoprotein I Antibodies Recognizes Epitopes in Domains 3, 4, and 5 That Are Located in a Lateral Zone of the Molecule (L-Shaped). Front. Immunol. 2019, 7, 1031.
- George, J.; Blank, M.; Levy, Y.; Meroni, P.; Damianovich, M.; Tincani, A.; Shoenfeld, Y. Differential effects of anti-beta2-glycoprotein I antibodies on endothelial cells and on the manifestations of experimental antiphospholipid syndrome. Circulation 1998, 97, 900–906.
- Sutjita, M.; Hohmann, A.; Comacchio, R.; Boey, M.L.; Bradley, J. A common anti-cardiolipin antibody idiotype in autoimmune disease: Identification using a mouse monoclonal antibody directed against a naturally-occurring anti-phospholipid antibody. Clin. Exp. Immunol. 1989, 75, 211–216.
- Blank, M.; Shoenfeld, Y.; Cabilly, S.; Heldman, Y.; Fridkin, M.; Katchalski-Katzir, E. Prevention of experimental antiphospholipid syndrome and endothelial cell activation by synthetic peptides. Proc. Natl. Acad. Sci. USA 1999, 96, 5164–5168.
- Agar, C.; Van Os, G.M.A.; Mörgelin, M.; Sprenger, R.R.; Marquart, J.A.; Urbanus, R.T.; Derksen, R.H.W.M.; Meijers, J.C.M.; De Groot, P.J. Beta2-glycoprotein I can exist in 2 conformations: Implications for our understanding of the antiphospholipid syndrome. Blood 2010, 116, 1336–1343.
- Buchholz, I.; Nestler, P.; Koppen, S.; Delcea, M. Lysine residues control the conformational dynamics of beta 2-glycoprotein I. Phys. Chem. Chem. Phys. 2018, 20, 26819–26829.
- Koike, T.; Ichikawa, K.; Kasahara, H.; Atsumi, T.; Tsutsumi, A.; Matsuura, E. Epitopes on beta2-GPI recognized by anticardiolipin antibodies. Lupus 1998, 7, 14–17.

