 +1 credit
+1 credit
Video Upload Options
Central pontine myelinolysis is a neurological condition involving severe damage to the myelin sheath of nerve cells in the pons (an area of the brainstem). It is predominately iatrogenic (treatment-induced), and is characterized by acute paralysis, dysphagia (difficulty swallowing), dysarthria (difficulty speaking), and other neurological symptoms. Central pontine myelinolysis was first described as a disorder in 1959. The original paper described four cases with fatal outcomes, and the findings on autopsy. The disease was described as a disease of alcoholics and malnutrition. ‘Central pontine’ indicated the site of the lesion and ‘myelinolysis’ was used to emphasise that myelin was affected. The authors intentionally avoided the term ‘demyelination’ to describe the condition, in order to differentiate this condition from multiple sclerosis and other neuroinflammatory disorders. Since this original description, demyelination in other areas of the central nervous system associated with osmotic stress has been described outside the pons (extrapontine). Osmotic demyelination syndrome is the term used for both central pontine myelinolysis and extrapontine myelinolysis. Central pontine myelinolysis, and osmotic demyelination syndrome, present most commonly as a complication of treatment of patients with profound hyponatremia (low sodium), which can result from a varied spectrum of conditions, based on different mechanisms. It occurs as a consequence of a rapid rise in serum tonicity following treatment in individuals with chronic, severe hyponatremia who have made intracellular adaptations to the prevailing hypotonicity.
1. Signs and Symptoms
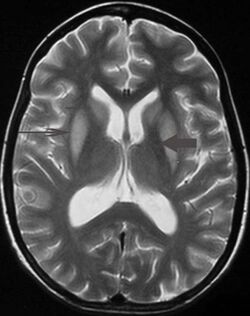
Symptoms depend on the regions of the brain involved. Prior to its onset, patients may present with the neurological signs and symptoms of hyponatraemic encephalopathy such as nausea and vomiting, confusion, headache and seizures. These symptoms may resolve with normalisation of the serum sodium concentration. Three to five days later, a second phase of neurological manifestations occurs correlating with the onset of myelinolysis. Observable immediate precursors may include seizures, disturbed consciousness, gait changes, and decrease or cessation of respiratory function.[1][2]
The classical clinical presentation is the progressive development of spastic quadriparesis, pseudobulbar palsy, and emotional lability (pseudobulbar affect), with other more variable neurological features associated with brainstem damage. These result from a rapid myelinolysis of the corticobulbar and corticospinal tracts in the brainstem.[3]
In about ten per cent of people with central pontine myelinolysis, extrapontine myelinolysis is also found. In these cases symptoms of Parkinson's disease may be generated.[4]
2. Causes
The most common cause is overly-rapid correction of low blood sodium levels (hyponatremia).[5] Apart from rapid correction of hyponatraemia, there are case reports of central pontine myelinolysis in association with hypokalaemia, anorexia nervosa when feeding is started, patients undergoing dialysis and burn victims. There is a case report of central pontine myelinolysis occurring in the context of refeeding syndrome, in the absence of hyponatremia.[6]
It has also been known to occur in patients suffering withdrawal symptoms of chronic alcoholism.[4] In these instances, occurrence may be entirely unrelated to hyponatremia or rapid correction of hyponatremia. It could affect patients who take some prescription medicines that are able to cross the blood-brain barrier and cause abnormal thirst reception - in this scenario the central pontine myelinolysis is caused by polydipsia leading to low blood sodium levels (hyponatremia).
In schizophrenic patients with psychogenic polydipsia, inadequate thirst reception leads to excessive water intake, severely diluting serum sodium.[7] With this excessive thirst combined with psychotic symptoms, brain damage such as central pontine myelinolysis[8] may result from hyperosmolarity caused by excess intake of fluids, (primary polydipsia) although this is difficult to determine because such patients are often institutionalised and have a long history of mental health conditions.[9]
It has been observed following hematopoietic stem cell transplantation.[10]
Central pontine myelinolysis may also occur in patients prone to hyponatremia affected by:
- Severe liver disease (e.g., cirrhosis)
- Liver transplant[11][12][13]
- Alcoholism
- Hypokalemia
- People with serum sodium <105 mEq/L
- Severe burns[14][15]
- Malnutrition
- Anorexia nervosa[16][17][18]
- Severe electrolyte disorders
- HIV/AIDS
- hyperemesis gravidarum[19][20]
- Hyponatremia due to peritoneal dialysis
- Wernicke encephalopathy[21]
3. Pathophysiology
The currently accepted theory states that the brain cells adjust their osmolarities by changing levels of certain osmolytes like inositol, betaine, and glutamine in response to varying serum osmolality. In the context of chronic low plasma sodium (hyponatremia), the brain compensates by decreasing the levels of these osmolytes within the cells, so that they can remain relatively isotonic with their surroundings and not absorb too much fluid. The reverse is true in hypernatremia, in which the cells increase their intracellular osmolytes so as not to lose too much fluid to the extracellular space.
With correction of the hyponatremia with intravenous fluids, the extracellular tonicity increases, followed by an increase in intracellular tonicity. When the correction is too rapid, not enough time is allowed for the brain's cells to adjust to the new tonicity, namely by increasing the intracellular osmoles mentioned earlier. If the serum sodium levels rise too rapidly, the increased extracellular tonicity will continue to drive water out of the brain's cells. This can lead to cellular dysfunction and central pontine myelinolysis.[22][23]
4. Diagnosis
It can be diagnosed clinically in the appropriate context, but may be difficult to confirm radiologically using conventional imaging techniques. Changes are more prominent on MRI than on CT, but often take days or weeks after acute symptom onset to develop. Imaging by MRI typically demonstrates areas of hyperintensity on T2-weighted images.[24]
5. Treatment
To minimise the risk of this condition developing from its most common cause, overly rapid reversal of hyponatremia, the hyponatremia should be corrected at a rate not exceeding 10 mmol/L/24 h or 0.5 mEq/L/h; or 18 mEq/L/48hrs; thus avoiding demyelination.[23] No large clinical trials have been performed to examine the efficacy of therapeutic re-lowering of serum sodium, or other interventions sometimes advocated such as steroids or plasma exchange.[23] Alcoholic patients should receive vitamin supplementation and a formal evaluation of their nutritional status.[25][26]
Once osmotic demyelination has begun, there is no cure or specific treatment. Care is mainly supportive. Alcoholics are usually given vitamins to correct for other deficiencies. The favourable factors contributing to the good outcome in central pontine myelinolysis without hyponatremia were: concurrent treatment of all electrolyte disturbances, early intensive care unit involvement at the advent of respiratory complications, early introduction of feeding including thiamine supplements with close monitoring of the electrolyte changes and input.[6]
Research has led to improved outcomes.[27] Animal studies suggest that inositol reduces the severity of osmotic demyelination syndrome if given before attempting to correct chronic hyponatraemia.[28] Further study is required before using inositol in humans for this purpose.[29]
6. Prognosis
Though traditionally the prognosis is considered poor, a good functional recovery is possible. All patients at risk of developing refeeding syndrome should have their electrolytes closely monitored, including sodium, potassium, magnesium, glucose and phosphate.[6] Recent data indicate that the prognosis of critically ill patients may even be better than what is generally considered,[30] despite severe initial clinical manifestations and a tendency by the intensivists to underestimate a possible favorable evolution.[31] While some patients die, most survive and of the survivors, approximately one-third recover; one-third are disabled but are able to live independently; one-third are severely disabled.[32] Permanent disabilities range from minor tremors and ataxia to signs of severe brain damage, such as spastic quadriparesis and locked-in syndrome.[33] Some improvements may be seen over the course of the first several months after the condition stabilizes.
The degree of recovery depends on the extent of the original axonal damage.[22]
References
- "Central pontine myelinolysis: case series and review". WMJ 104 (6): 56–60. August 2005. PMID 16218318. http://www.ncbi.nlm.nih.gov/pubmed/16218318
- "Central pontine and extrapontine myelinolysis: from epileptic and other manifestations to cognitive prognosis". Journal of Neurology 257 (7): 1176–1180. July 2010. doi:10.1007/s00415-010-5486-7. PMID 20148334. https://dx.doi.org/10.1007%2Fs00415-010-5486-7
- "Pontine and extrapontine myelinolysis: a neurologic disorder following rapid correction of hyponatremia". Medicine 72 (6): 359–373. November 1993. doi:10.1097/00005792-199311000-00001. PMID 8231786. https://dx.doi.org/10.1097%2F00005792-199311000-00001
- "Central pontine and extrapontine myelinolysis after alcohol withdrawal". Alcohol and Alcoholism 43 (6): 647–649. 2008. doi:10.1093/alcalc/agn050. PMID 18678596. https://dx.doi.org/10.1093%2Falcalc%2Fagn050
- "Improvement of central pontine myelinolysis as demonstrated by repeated magnetic resonance imaging in a patient without evidence of hyponatremia". Acta Neurologica Belgica 99 (3): 189–193. September 1999. PMID 10544728. http://www.ncbi.nlm.nih.gov/pubmed/10544728
- "Central pontine myelinolysis without hyponatraemia". The Journal of the Royal College of Physicians of Edinburgh 41 (3): 211–214. September 2011. doi:10.4997/JRCPE.2011.305. PMID 21949915. https://dx.doi.org/10.4997%2FJRCPE.2011.305
- "Psychogenic Polydipsia (Excessive Fluid seeking Behaviour)". American Psychological Society Divisions. http://www.apadivisions.org/division-31/publications/articles/british-columbia/psychogenic-polydipsia.pdf.
- "Psychotic disorder in a patient with central and extrapontine myelinolysis". Psychiatry and Clinical Neurosciences 61 (3): 320–322. June 2007. doi:10.1111/j.1440-1819.2007.01648.x. PMID 17472602. https://dx.doi.org/10.1111%2Fj.1440-1819.2007.01648.x
- "Psychogenic polydipsia: the result, or cause of, deteriorating psychotic symptoms? A case report of the consequences of water intoxication". Case Reports in Psychiatry 2015: 846459. 2015-01-21. doi:10.1155/2015/846459. PMID 25688318. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4320790
- "Central pontine myelinolysis in a patient with acute lymphoblastic leukemia after hematopoietic stem cell transplantation: a case report". Journal of Korean Medical Science 23 (2): 324–327. April 2008. doi:10.3346/jkms.2008.23.2.324. PMID 18437020. PMC 2526450. http://jkms.org/contents/jkms.php?pubyear=2008&vol=23&fpage=324.
- "Central nervous system lesions in adult liver transplant recipients: clinical review with implications for management". Medicine 73 (2): 110–118. March 1994. doi:10.1097/00005792-199403000-00004. PMID 8152365. https://dx.doi.org/10.1097%2F00005792-199403000-00004
- "Subclinical central pontine myelinolysis following liver transplantation". Brain & Development 24 (3): 179–182. April 2002. doi:10.1016/S0387-7604(02)00013-X. PMID 11934516. https://dx.doi.org/10.1016%2FS0387-7604%2802%2900013-X
- "Neurologic complications of liver transplantation". Neurologic Clinics 6 (2): 327–348. May 1988. doi:10.1016/S0733-8619(18)30873-9. PMID 3047544. https://dx.doi.org/10.1016%2FS0733-8619%2818%2930873-9
- "Central pontine myelinolysis in severely burned patients: relationship to serum hyperosmolality". Neurology 38 (8): 1211–1217. August 1988. doi:10.1212/wnl.38.8.1211. PMID 3399069. https://dx.doi.org/10.1212%2Fwnl.38.8.1211
- "Central nervous system complications of thermal burns. A postmortem study of 139 patients". Medicine 71 (5): 271–283. September 1992. doi:10.1097/00005792-199209000-00002. PMID 1522803. https://dx.doi.org/10.1097%2F00005792-199209000-00002
- "Central pontine myelinolysis associated with hypokalaemia in anorexia nervosa". Journal of Neurology, Neurosurgery, and Psychiatry 74 (3): 353–355. March 2003. doi:10.1136/jnnp.74.3.353. PMID 12588925. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1738317
- "Central pontine myelinolysis associated with hypokalaemia in anorexia nervosa". Journal of Neurology, Neurosurgery, and Psychiatry 75 (4): 663; author reply 663. April 2004. PMID 15026526. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1739009
- "Centropontine myelinolysis related to refeeding syndrome in an adolescent suffering from anorexia nervosa". Neuropediatrics 43 (3): 152–154. June 2012. doi:10.1055/s-0032-1307458. PMID 22473289. https://dx.doi.org/10.1055%2Fs-0032-1307458
- "Wernicke's encephalopathy and central pontine myelinolysis associated with hyperemesis gravidarum". BMJ 305 (6852): 517–518. August 1992. doi:10.1136/bmj.305.6852.517. PMID 1393001. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1882865
- "Wernicke's encephalopathy and central pontine myelinolysis in hyperemesis gravidarum". Journal of Neurosciences in Rural Practice 4 (1): 39–41. January 2013. doi:10.4103/0976-3147.105608. PMID 23546346. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3579041
- "Rapid development of central pontine myelinolysis after recovery from Wernicke encephalopathy: a non-alcoholic case without hyponatremia". Internal Medicine 51 (12): 1599–1603. 2012. doi:10.2169/internalmedicine.51.7498. PMID 22728498. https://dx.doi.org/10.2169%2Finternalmedicine.51.7498
- "Axonal damage: a key predictor of outcome in human CNS diseases". Brain 126 (Pt 3): 515–530. March 2003. doi:10.1093/brain/awg061. PMID 12566274. https://dx.doi.org/10.1093%2Fbrain%2Fawg061
- "Clinical practice guideline on diagnosis and treatment of hyponatraemia". European Journal of Endocrinology 170 (3): G1-47. March 2014. doi:10.1530/eje-13-1020. PMID 24569125. https://dx.doi.org/10.1530%2Feje-13-1020
- "Intracranial Lesions with Low Signal Intensity on T2-weighted MR Images - Review of Pathologies". Polish Journal of Radiology 80: 40–50. 2015. doi:10.12659/PJR.892146. PMID 25628772. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4307690
- "Rapid correction of hyponatremia causes demyelination: relation to central pontine myelinolysis". Science 211 (4486): 1068–1070. March 1981. doi:10.1126/science.7466381. PMID 7466381. Bibcode: 1981Sci...211.1068K. https://dx.doi.org/10.1126%2Fscience.7466381
- "Experimental pontine and extrapontine myelinolysis". Transactions of the American Neurological Association 105: 354–358. 1980. PMID 7348981. http://www.ncbi.nlm.nih.gov/pubmed/7348981
- "Osmotic demyelination disorders: central pontine and extrapontine myelinolysis". Current Opinion in Neurology 13 (6): 691–697. December 2000. doi:10.1097/00019052-200012000-00014. PMID 11148672. https://dx.doi.org/10.1097%2F00019052-200012000-00014
- "Myoinositol administration improves survival and reduces myelinolysis after rapid correction of chronic hyponatremia in rats". Journal of Neuropathology and Experimental Neurology 65 (1): 37–44. January 2006. doi:10.1097/01.jnen.0000195938.02292.39. PMID 16410747. https://dx.doi.org/10.1097%2F01.jnen.0000195938.02292.39
- "INOSITOL: Overview, Uses, Side Effects, Precautions, Interactions, Dosing and Reviews" (in en). https://www.webmd.com/vitamins/ai/ingredientmono-299/inositol.
- "Long-term outcome of patients hospitalized in intensive care units with central or extrapontine myelinolysis*". Critical Care Medicine 40 (3): 970–972. March 2012. doi:10.1097/CCM.0b013e318236f152. PMID 22036854. https://dx.doi.org/10.1097%2FCCM.0b013e318236f152
- "Central pontine myelinolysis: a lesson in humility*". Critical Care Medicine 40 (3): 1026–1027. March 2012. doi:10.1097/CCM.0b013e31823b8e0b. PMID 22343870. https://dx.doi.org/10.1097%2FCCM.0b013e31823b8e0b
- "Osmotic demyelination syndrome". BMJ 331 (7520): 829–830. October 2005. doi:10.1136/bmj.331.7520.829. PMID 16210283. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1246086
- "Central Pontine Myelinolysis". 17 November 2015. http://emedicine.medscape.com/article/1174329-overview.


