 +1 credit
+1 credit
Video Upload Options
Live cell imaging is the study of living cells using time-lapse microscopy. It is used by scientists to obtain a better understanding of biological function through the study of cellular dynamics. Live cell imaging was pioneered in first decade of the 21th century. One of the first time-lapse microcinematographic films of cells ever made was made by Julius Ries, showing the fertilization and development of the sea urchin egg. Since then, several microscopy methods have been developed which allow researchers to study living cells in greater detail with less effort. A newer type of imaging utilizing quantum dots have been used as they are shown to be more stable. The development of holotomographic microscopy has disregarded phototoxicity and other staining-derived disadvantages by implementing digital staining based on cells’ refractive index.
1. Overview
Biological systems exist as a complex interplay of countless cellular components interacting across four dimensions to produce the phenomenon called life. While it is common to reduce living organisms to non-living samples to accommodate traditional static imaging tools, the further the sample deviates from the native conditions the more likely the delicate processes in question will exhibit perturbations.[1] The onerous task of capturing the true physiological identity of living tissue, therefore, requires high-resolution visualization across both space and time within the parent organism.[2] The technological advances of live-cell imaging, designed to provide spatiotemporal images of subcellular events in real-time, serves an important role for corroborating the biological relevance of physiological changes observed during experimentation. Due to their contiguous relationship with physiological conditions, live-cell assays are considered the standard for probing complex and dynamic cellular events.[3] As dynamic processes such as migration, cell development, and intracellular trafficking increasingly become the focus of biological research, techniques capable of capturing 3-dimensional data in real-time for cellular networks (in situ) and entire organisms (in vivo) will become indispensable tools in understanding biological systems. The general acceptance of live-cell imaging has led to a rapid expansion in the number of practitioners and established a need for increased spatial and temporal resolution without compromising the health of the cell.[4]
2. Types of Microscopy Used
2.1. Phase Contrast
Before the introduction of the phase contrast microscope it was difficult to observe living cells. As living cells are translucent they must be stained to be visible in a traditional light microscope. Unfortunately, the process of staining cells generally kills the cells. With the invention of the phase contrast microscopy it became possible to observe unstained living cells in detail. After its introduction in the 1940s, live cell imaging rapidly became popular using phase contrast microscopy.[5] The phase contrast microscope was popularized through a series of time-lapse movies (Video 1), recorded using a photographic film camera.[6] Its inventor, Frits Zernike, was awarded the Nobel Prize in 1953.[7] Other later phase contrast techniques used to observe unstained cells are Hoffman modulation and differential interference contrast microscopy.
2.2. Fluorescent
Phase contrast microscopy does not have the capacity to observe specific proteins or other organic chemical compounds which form the complex machinery of a cell. Synthetic and organic fluorescent stains have therefore been developed to label such compounds, making them observable by fluorescent microscopy (Video 2).[8] Fluorescent stains are, however, phototoxic, invasive and bleach when observed. This limits their use when observing living cells over extended periods of time. Non-invasive phase contrast techniques are therefore often used as a vital complement to fluorescent microscopy in live cell imaging applications.[9][10]
2.3. Quantitative Phase Contrast
As a result of the rapid increase in pixel density of digital image sensors, quantitative phase contrast microscopy has emerged as an alternative microscopy method for live cell imaging.[11][12] Quantitative phase contrast microscopy has an advantage over fluorescent and phase contrast microscopy in that it is both non-invasive and quantitative in its nature.
Due to the narrow focal depth of conventional microscopy, live cell imaging is to a large extent currently limited to observing cells on a single plane. Most implementations of quantitative phase contrast microscopy allow for images to be created and focused at different focal planes from a single exposure. This opens up the future possibility of 3-dimensional live cell imaging by means of fluorescence techniques.[13] Quantitative phase contrast microscopy with rotational scanning allow 3D time-lapse images of living cells to be acquired at high resolution.[14][15][16]
2.4. Holotomography
Holotomography (HT) is a laser technique to measure three-dimensional refractive index (RI) tomogram of a microscopic sample such as biological cells and tissues. Because the RI can serve as an intrinsic imaging contrast for transparent or phase objects, measurements of RI tomograms can provide label-free quantitative imaging of microscopic phase objects. In order to measure 3-D RI tomogram of samples, HT employs the principle of holographic imaging and inverse scattering. Typically, multiple 2D holographic images of a sample are measured at various illumination angles, employing the principle of interferometric imaging. Then, a 3D RI tomogram of the sample is reconstructed from these multiple 2D holographic images by inversely solving light scattering in the sample.
The principle of HT is very similar to X-ray computed tomography (CT) or CT scan. CT scan measures multiple 2-D X-ray images of a human body at various illumination angles, and a 3-D tomogram (X-ray absorptivity) is then retrieved via the inverse scattering theory. Both the X-ray CT and laser HT shares the same governing equation – Helmholtz equation, the wave equation for a monochromatic wavelength. HT is also known as optical diffraction tomography.
The combination of holography and rotational scanning allows for long-term, label-free, live cell recordings.
Non-invasive optical nanoscopy can achieve such a lateral resolution by using a quasi-2π-holographic detection scheme and complex deconvolution. The spatial frequencies of the imaged cell do not make any sense to the human eye. But these scattered frequencies are converted into a hologram and synthesize a bandpass which has a resolution double the one normally available. Holograms are recorded from different illumination directions on the sample plane and observe sub-wavelength tomographic variations of the specimen. Nanoscale apertures serve to calibrate the tomographic reconstruction and to characterize the imaging system by means of the coherent transfer function. This gives rise to realistic inverse filtering and guarantees true complex field reconstruction.[17]
In conclusion, the 2 terminologies of (i) optical resolution (the real one) and (ii) sampling resolution (the one on the screen) are separated for 3D holotomographic microscopy.
3. Instrumentation and Optics
Live-cell imaging represents a careful compromise between acquiring the highest-resolution image and keeping the cells alive for as long as possible.[18] As a result, live-cell microscopists face a unique set of challenges that are often overlooked when working with fixed-specimens. Moreover, live-cell imaging often employs special optical system and detector specifications. For example, ideally the microscopes used in live-cell imaging would have high signal-to-noise ratios, fast image acquisition rates to capture time-lapse video of extracellular events, and maintaining the long-term viability of the cells.[19] However, optimizing even a single facet of image acquisition can be resource intensive and should be considered on a case by case basis.
3.1. Lens Designs
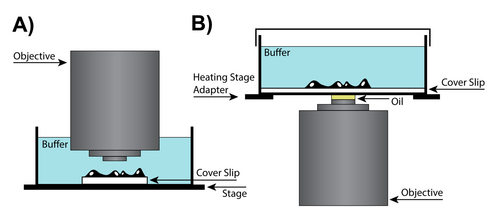
Low magnification "dry"
In cases where extra space between the objective and the specimen is required to work with the sample, a dry lens can be used, potentially requiring additional adjustments of the correction collar, which changes the location of the lens in the objective, to account for differences in imaging chambers. Special objective lenses are designed with correction collars that correct for spherical aberrations while accounting for the cover slip thickness. In high numerical aperture (NA) dry objective lenses, the correction collar adjustment ring will change the position of a movable lens group to account for differences in the way the outside of the lens focuses light relative to the center. Although lens aberrations are inherent in all lens designs, they become more problematic in dry lenses where resolution retention is key.[20]
Oil immersion high NA
Oil immersion is a technique that can increase image resolution by immersing the lens and the specimen in oil with a high refractive index. Since light bends when it passes between mediums with different refractive indexes, by placing oil with the same refractive index as glass between the lens and the slide, two transitions between refractive indices can be avoided.[21] However, for most applications it is recommended that oil immersion be used with fixed (dead) specimens because live cells require an aqueous environment and the mixing of oil and water can cause severe spherical aberrations. For some applications silicone oil can be used to produce more accurate image reconstructions. Silicone oil is an attractive media because it has a refractive index that is close to that of living cells, allowing it to produce high resolution images while minimizing spherical aberrations.[20]
Water immersion
Live-cell imaging requires a sample in an aqueous environment that is often 50 to 200 micrometers away from the cover glass. Therefore, water immersion lenses can help achieve a higher resolving power due to the fact that both the environment and the cells themselves will be close to the refractive index of water. Water immersion lenses are designed to be compatible with the refractive index of water and usually have a corrective collar which allows for adjustment of the objective. Additionally, because of the higher refractive index of water, water immersion lenses have a high numerical aperture and can produce images superior to oil immersion lens when resolving planes deeper than 0 µm.[20]
Dipping
Another solution for live-cell imaging is the dipping lens. These lenses are a subset of water immersion lenses that do not require a cover slip and can be dipped directly into the aqueous environment of the sample. One of the main advantages of the dipping lens is that it has a long effective working distance.[22] Since a cover slip is not required, this type of lens can approach the surface of the specimen and as a result, the resolution is limited by the restraints imposed by spherical aberration rather than the physical limitations of the cover slip. Although dipping lenses can be very useful, they are not ideal for all experiments since the act of "dipping" the lens can disturb the cells in the sample. Additionally, since the incubation chamber must be open to the lens, changes in the sample environment due to evaporation must be closely monitored.[20]
4. Phototoxicity and Photobleaching
Today, most live imaging techniques rely on either high illumination regimes or fluorescent labelling, both inducing phototoxicity and compromising the ability to keep cells unperturbed and alive over time. Since our knowledge of biology is driven by observation, it is key to minimize the perturbations induced by the imaging technique.
The rise of confocal microscopy is closely correlated with accessibility of high power lasers, which are able to achieve high intensities of light excitation. However, the high power output can damage sensitive fluorophores and are usually run significantly below their maximum power output.[23] Over exposure to light can result in photodamage due to photobleaching or phototoxicity. The effects of photobleaching can significantly reduce the quality of fluorescent images and in recent years there has been a significant demand for longer-lasting commercial fluorophores. One solution, the Alexa Fluor series, show little to no fading even at high laser intensities.[24]
Under physiological conditions, many cells and tissue types are exposed to only low levels of light.[25] As a result, it is import to minimize the exposure of live cells to high doses of ultraviolet (UV), infrared (IR), or fluorescence exciting wavelengths of light, which can damage DNA, raise cellular temperatures, and cause photo bleaching respectively.[26] High energy photons absorbed by the fluorophores and the sample are emitted at longer wavelengths proportional to the Stokes shift.[27] However, cellular organelles can be damaged when the photon's energy produces chemical and molecular changes rather than being re-emitted.[28] It is believed that the primary culprit in the light induced toxicity experienced by live cells is a result of free radicals produced by the excitation of fluorescent molecules.[25] These free radicals are highly reactive and will result in the destruction of cellular components, which can result in non-physiological behavior.
One method of minimizing photo-damage is to lower the oxygen concentration in the sample to avoid the formation of reactive oxygen species.[29] However, this method is not always possible in live-cell imaging and may require additional intervention. Another method for reducing the effects of free radicals in the sample is the use of antifade reagents. Unfortunately, most commercial antifade reagents cannot be used in live-cell imaging because of their toxicity.[30] Instead, natural free-radical scavengers such as vitamin C or vitamin E can be used without substantially altering physiological behavior on shorter time scales.[31] Phototoxicity-free live cell imaging has recently been developed and commercialised. Holotomographic microscopy avoids phototoxicity thanks to its low power laser (Laser class 1: 0.2 mW/mm2).[32][33][34]
References
- Petroll, W. M.; Jester, J. V.; Cavanagh, H. D. (May 1994). "In vivo confocal imaging: general principles and applications". Scanning 16 (3): 131–149. ISSN 0161-0457. PMID 8038913. http://www.worldcat.org/issn/0161-0457
- Meijering, Erik; Dzyubachyk, Oleh; Smal, Ihor (2012-01-01). Methods for Cell and Particle Tracking. 504. 183–200. doi:10.1016/B978-0-12-391857-4.00009-4. ISBN 9780123918574. https://zenodo.org/record/3414054.
- Allan, Victoria J.; Stephens, David J. (2003-04-04). "Light Microscopy Techniques for Live Cell Imaging". Science 300 (5616): 82–86. doi:10.1126/science.1082160. ISSN 1095-9203. PMID 12677057. Bibcode: 2003Sci...300...82S. https://dx.doi.org/10.1126%2Fscience.1082160
- DanceMar. 27, Amber; 2018; Pm, 2:10 (2018-03-27). "Live-cell imaging: Deeper, faster, wider". https://www.sciencemag.org/features/2018/03/live-cell-imaging-deeper-faster-wider.
- Burgess, Mark (15 October 2003). "Celebrating 50 years of Live Cell Imaging". London: The Biochemical Society. http://www.biochemist.org/bio/02506/0046/025060046.pdf.
- Gundlach H. "50 Years Ago: Frits Zernike (1888-1966) Got the Nobel Prize in Physics for the Development of the Phase Contrast Method" (PDF) (Press release). Carl Zeiss AG. Archived from the original (PDF) on March 22, 2014. https://web.archive.org/web/20140322045926/http://www.zeiss.com/C1256F8500454979/0/B9369EB45E3860A0C1256F8E003AC5DC/%24file/frits-zernike-dgz_e.pdf
- "The Nobel Prize in Physics 1953". Nobel Media AB. https://www.nobelprize.org/nobel_prizes/physics/laureates/1953/.
- Stockert, Juan Carlos; Blázquez-Castro, Alfonso (2017). Fluorescence Microscopy in Life Sciences. Bentham Science Publishers. ISBN 978-1-68108-519-7. https://ebooks.benthamscience.com/book/9781681085180/. Retrieved 24 December 2017.
- "Light microscopy techniques for live cell imaging". Science 300 (5616): 82–6. April 2003. doi:10.1126/science.1082160. PMID 12677057. Bibcode: 2003Sci...300...82S. https://dx.doi.org/10.1126%2Fscience.1082160
- "Standard fluorescent imaging of live cells is highly genotoxic". Cytometry. Part A 83 (6): 552–60. June 2013. doi:10.1002/cyto.a.22291. PMID 23650257. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3677558
- "Quantitative phase imaging in biomedicine". Nature Photonics 12 (10): 578–589. 2018. doi:10.1038/s41566-018-0253-x. PMID 26648557. Bibcode: 2018NaPho..12..578P. https://dx.doi.org/10.1038%2Fs41566-018-0253-x
- Cuche, Etienne; Bevilacqua, Frédéric; Depeursinge, Christian (1999). "Digital holography for quantitative phase-contrast imaging". Optics Letters 24 (5): 291–293. doi:10.1364/OL.24.000291. PMID 18071483. Bibcode: 1999OptL...24..291C. https://semanticscholar.org/paper/abc6cc4fe12c8dac17ac83e19f172c7a991ae824.
- Rosen, Joseph; Brooker, Gary (2008). "Non-scanning motionless fluorescence three-dimensional holographic microscopy". Nature Photonics 2 (3): 190–195. doi:10.1038/nphoton.2007.300. Bibcode: 2008NaPho...2..190R. https://semanticscholar.org/paper/9608943ab5b2aa8b6f911a6e78d02370a0b8b1f1.
- "Tomographic phase microscopy". Nature Methods 4 (9): 717–719. 2007. doi:10.1038/nmeth1078. PMID 17694065. https://dx.doi.org/10.1038%2Fnmeth1078
- "Marker-free phase nanoscopy". Nature Photonics 7 (2): 113–117. 2013. doi:10.1038/nphoton.2012.329. Bibcode: 2013NaPho...7..113C. https://semanticscholar.org/paper/1d5421d2147c5bc27e6f73b8c09ae85b39072133.
- "Stain-free 3D Nanoscopy of Living Cells". Optik & Photonik (Wiley Online Library) 11: 38–42. 2016. doi:10.1002/opph.201600008. https://dx.doi.org/10.1002%2Fopph.201600008
- Cotte, Yann; Toy, Fatih; Jourdain, Pascal; Pavillon, Nicolas; Boss, Daniel; Magistretti, Pierre; Marquet, Pierre; Depeursinge, Christian (February 2013). "Marker-free phase nanoscopy". Nature Photonics 7 (2): 113–117. doi:10.1038/nphoton.2012.329. ISSN 1749-4893. Bibcode: 2013NaPho...7..113C. https://www.nature.com/articles/nphoton.2012.329.
- "Overview of live-cell imaging: requirements and methods used". Anatomical Record 296 (1): 1–8. January 2013. doi:10.1002/ar.22554. PMID 22907880. https://dx.doi.org/10.1002%2Far.22554
- "Live-cell fluorescence imaging". Digital Microscopy. 114. 2013. 125–50. doi:10.1016/B978-0-12-407761-4.00006-3. ISBN 9780124077614. https://dx.doi.org/10.1016%2FB978-0-12-407761-4.00006-3
- Hibbs, Albert R (2004). Confocal microscopy for biologists. New York: Kluwer Academic/Plenum Publishers. ISBN 978-0306484681. OCLC 54424872. http://www.worldcat.org/oclc/54424872
- "Solid immersion microscope". Applied Physics Letters 57 (24): 2615–2616. 1990-12-10. doi:10.1063/1.103828. Bibcode: 1990ApPhL..57.2615M. https://dx.doi.org/10.1063%2F1.103828
- "Objective Lenses for Confocal Microscopy", Handbook of Biological Confocal Microscopy, Springer US, 2006, pp. 145–161, doi:10.1007/978-0-387-45524-2_7, ISBN 9780387259215, https://semanticscholar.org/paper/9935a9c37c93ddb34a3c554f83e451a4a1db6d4f
- Amos, W.B.; White, J.G. (2003-09-01). "How the Confocal Laser Scanning Microscope entered Biological Research". Biology of the Cell 95 (6): 335–342. doi:10.1016/S0248-4900(03)00078-9. PMID 14519550. https://dx.doi.org/10.1016%2FS0248-4900%2803%2900078-9
- "Improved fluoroimmunoassays using the dye Alexa Fluor 647 with the RAPTOR, a fiber optic biosensor 7". Journal of Immunological Methods 271 (1–2): 17–24. 2002-12-20. doi:10.1016/S0022-1759(02)00327-7. ISSN 0022-1759. PMID 12445725. https://dx.doi.org/10.1016%2FS0022-1759%2802%2900327-7
- "Live-cell microscopy - tips and tools". Journal of Cell Science 122 (Pt 6): 753–67. March 2009. doi:10.1242/jcs.033837. PMID 19261845. https://dx.doi.org/10.1242%2Fjcs.033837
- "Circumventing photodamage in live-cell microscopy". Digital Microscopy. 114. 2013. 545–60. doi:10.1016/B978-0-12-407761-4.00023-3. ISBN 9780124077614. https://dx.doi.org/10.1016%2FB978-0-12-407761-4.00023-3
- Rost, Fred W (1992–1995). Fluorescence microscopy. Cambridge: Cambridge University Press. ISBN 978-0521236416. OCLC 23766227. http://www.worldcat.org/oclc/23766227
- "Assessing phototoxicity in live fluorescence imaging". Nature Methods 14 (7): 657–661. June 2017. doi:10.1038/nmeth.4344. PMID 28661494. https://dx.doi.org/10.1038%2Fnmeth.4344
- "Fluorescence live cell imaging". Quantitative Imaging in Cell Biology. 123. 2014. 77–94. doi:10.1016/B978-0-12-420138-5.00005-7. ISBN 9780124201385. https://dx.doi.org/10.1016%2FB978-0-12-420138-5.00005-7
- Pawley, James B (2006). Handbook of biological confocal microscopy (3rd ed.). New York, NY: Springer. ISBN 9780387455242. OCLC 663880901. http://www.worldcat.org/oclc/663880901
- Watu, Aswani; Metussin, Nurzaidah; Yasin, Hartini M.; Usman, Anwar (2018). "The total antioxidant capacity and fluorescence imaging of selected plant leaves commonly consumed in Brunei Darussalam". AIP Conference Proceedings 1933 (1): 020001. doi:10.1063/1.5023935. Bibcode: 2018AIPC.1933b0001W. https://dx.doi.org/10.1063%2F1.5023935
- Pollaro, L.; Equis, S.; Dalla Piazza, B.; Cotte, Y. (2016). "Stain‐free 3D Nanoscopy of Living Cells". Optik & Photonik 11: 38–42. doi:10.1002/opph.201600008. https://dx.doi.org/10.1002%2Fopph.201600008
- Pollaro, L.; Dalla Piazza, B.; Cotte, Y. (2015). "Digital Staining: Microscopy of Live Cells Without Invasive Chemicals". Microscopy Today 23 (4): 12–17. doi:10.1017/S1551929515000590. http://doc.rero.ch/record/301087/files/S1551929515000590.pdf.
- Patrick A. Sandoz, Christopher Tremblay, Sebastien Equis, Sorin Pop, Lisa Pollaro, YannCotte, F. Gisou van der Goot, Mathieu Frechin. Label free 3D analysis of organelles in living cells by refractive index shows pre-mitotic organelle spinning in mammalian stem cells bioRxiv 407239; doi: https://doi.org/10.1101/407239
- Pollaro, L.; Equis, S.; Dalla Piazza, B.; Cotte, Y. (2016). "Stain‐free 3D Nanoscopy of Living Cells". Optik & Photonik 11: 38–42. doi:10.1002/opph.201600008. https://dx.doi.org/10.1002%2Fopph.201600008
- Pollaro, L.; Dalla Piazza, B.; Cotte, Y. (2015). "Digital Staining: Microscopy of Live Cells Without Invasive Chemicals". Microscopy Today 23 (4): 12–17. doi:10.1017/S1551929515000590. http://doc.rero.ch/record/301087/files/S1551929515000590.pdf.
- Patrick A. Sandoz, Christopher Tremblay, Sebastien Equis, Sorin Pop, Lisa Pollaro, YannCotte, F. Gisou van der Goot, Mathieu Frechin. Label free 3D analysis of organelles in living cells by refractive index shows pre-mitotic organelle spinning in mammalian stem cells bioRxiv 407239; doi: https://doi.org/10.1101/407239



