Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhu, H.; Yan, Y.; Jiang, Y.; Meng, X. Effects of Ellagic Acid in Neurological Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/31180 (accessed on 24 July 2026).
Zhu H, Yan Y, Jiang Y, Meng X. Effects of Ellagic Acid in Neurological Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/31180. Accessed July 24, 2026.
Zhu, Heyu, Yuanmei Yan, Yi Jiang, Xianfang Meng. "Effects of Ellagic Acid in Neurological Diseases" Encyclopedia, https://encyclopedia.pub/entry/31180 (accessed July 24, 2026).
Zhu, H., Yan, Y., Jiang, Y., & Meng, X. (2022, October 25). Effects of Ellagic Acid in Neurological Diseases. In Encyclopedia. https://encyclopedia.pub/entry/31180
Zhu, Heyu, et al. "Effects of Ellagic Acid in Neurological Diseases." Encyclopedia. Web. 25 October, 2022.
Copy Citation
Aging is associated with several diseases that threaten the health of older people, such as neurodegenerative diseases, which cause indelible and irreversible damage to both the physical and psychological well-being of older people and impose a heavy burden on their families and society. In recent years, a large number of studies have focused on the therapeutic effects of Ellagic Acid, demonstrating the health benefits of EA for neurodegenerative diseases. Three common neurological diseases are described here: AD, PD, and cerebral ischemia as well as the antioxidative and anti-inflammatory protective effects of EA.
ellagic acid
anti-aging
anti-inflammation
1. EA in Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases around the world, especially in the aging population. The patients behave with cognitive disorders, aphasia, and personality changes, which seriously affect patients’ quality of life [1]. The pathological change of AD can be concluded as three main features: extracellular amyloid-beta (Aβ) deposition, intracellular neurofibrillary tangles (NFT, caused by hyperphosphorylation of tau protein), and the loss of synapses and neurons.
Oxidative imbalance plays an important role in the pathogenesis of AD. The sources of free radicals in AD can be summarized in three aspects: (1) the accumulation of Fe and Al in neurons containing NFT can lead to the formation of OH·; (2) activated microglia that can form NO and O−2·; and (3), the abnormal metabolism of mitochondria can produce ROS [2]. It has been found that Fe, Zn, and Cu can bind to Aβ and APP and increase the oxidative stress of the AD brain [3]. Another pathologic feature of AD is the hyperphosphorylation of τ proteins, where the phosphorylation process is associated with the activation and oxidation of Nf-κB; this is important because the τ protein may be related to oxidative stress [4]. It has been clarified that EA can reduce the death of cells containing Aβ, lower the production of ROS, and mitigate DNA damage [5]. With its powerful free radical scavenging ability, EA can inhibit lipid peroxidation and reduce the production of NO, glycation end products, and so on [6].
In recent years, much evidence has proven that inflammation plays an important role in AD pathogenesis with the pathological observations of the inflammatory process [7]. As one of the pathogenetic features of AD, Aβ has been proven to activate the complement system and microglia [1]. Microglia are the immune cells in the CNS and the activation of microglia leads to the secretion of inflammatory cytokines, anaphylatoxin, free radicals, and neurotoxic substances which can kill neurons [1][8][9]. A recent study has shown that the activated microglia can synthesize proteolytic enzymes which can destroy cell structures and lead to the dysfunction of neurons [10]. The T-cells can also be activated in such a process and release inflammatory cytokines such as IL-1α, IL-1β, and TNF-α [11]. The excessive release of inflammatory cytokines induces the production of APP, which will positively upregulate the production of Aβ [9]. The continuous activation of pro-inflammatory responses could cause damage to the brain and the worsening of AD [10]. A study revealed that EA can regulate the production of IL-1β and TNF-1α to protect the damaged neurons induced by arsenic and improve the behavior and pathological change of AD [12]. Another key finding indicates that EA can reduce the number of microglia and inhibit the production of inflammatory cytokines [13] (Figure 1).
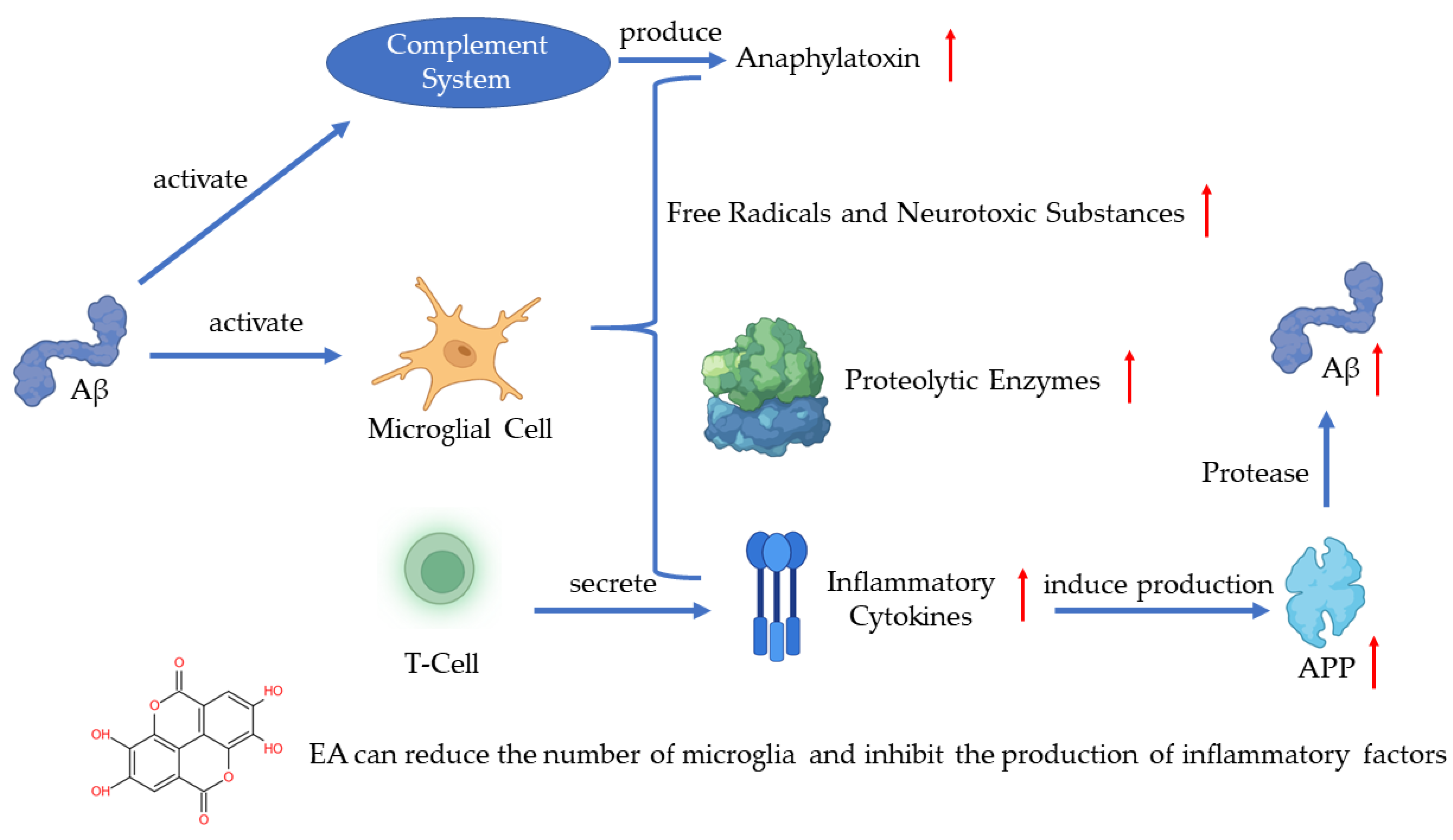
Figure 1. Pathogenesis of AD and the role of EA.
Plenty of studies focused on the Aβ and τ protein have shown the effects of EA. Researchers found that a group treated with EA exhibited a reduced level of Aβ plaques [14][15] and EA can significantly inhibit the hyperphosphorylation of the τ protein in animals’ hippocampus [15]. As a natural substance, EA is a potential therapeutic prospect for AD treatment.
2. EA in Parkinson’s Disease
PD is recognized as the most common neurodegenerative disorder after AD. The motor features of PD include bradykinesia, muscular rigidity, and resting tremors. The non-motor features include olfactory dysfunction, cognitive impairment, psychiatric symptoms, and autonomic dysfunction. The specific degeneration of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies (LB) have been recognized microscopically [16]. It has been proven that oxidative stress is one of the primary underlying reasons for substantia nigra pars compacta loss, and neuroinflammation has been recognized as a culprit contributing to PD development [17].
More and more evidence indicates that oxidative stress is a key driver of dopaminergic neurodegeneration in all forms of PD [18][19] and ROS accumulation results in damage to the structural integrity and neuronal dysfunction [20]. Moreover, several epidemiological studies indicate that the use of anti-inflammatory medications reduces the risk of PD [21]. Other studies also revealed that inflammation can directly or indirectly contribute to the development and progression of PD [22]. There is a vicious circle between PD, redox imbalance, and chronic inflammation. As people age, excessive oxidative stress or brain injury can lead to microglial activation [23], and the activated microglia secrete pro-inflammatory factors such as TNF-α, IL-1β, and IL-18 [24]. The accumulation of pro-inflammatory factors leads to the continuing loss of dopamine neurons. The damaged dopamine neurons release neurotoxic factors which induce a secondary activation of microglia that further secrete pro-inflammatory factors leading to more loss of dopamine neurons, thus creating a vicious cycle leading to neuroinflammation and neurodegeneration [25].
Several studies have shown that EA plays an active role in PD by enhancing the antioxidant defense and reducing oxidative stress [26][27][28]. EA can reduce oxidative damage by regulating the Nrf2 and Nf-κB pathways and improving the activity of antioxidant enzymes and antioxidants [29]. It has also been indicated that EA can lead to a decreased malondialdehyde (MDA) level and increased activities of total glutathione (GSH), catalase, and SOD in the mouse model of PD [26]. Pretreatment with EA in the PD rats reduced ROS and improved the level of monoamine oxidase B (MAO-B), Nrf2, and HO-1. Additionally, after using the antagonist, the improved results disappeared [27]. In another study, after the oral administration of EA, the medial forebrain bundle (MFB)-injured rats appeared to recover their motor deficiencies by a significant increase in glutathione peroxidase (GPx) and SOD activity and decreasing the level of MDA [28]. The above evidence has shown that EA can protect dopamine neurons by inhibiting oxidative stress in the animal model of PD.
EA can also inhibit the release of endogenous inflammatory mediators in the animal model of PD, such as COX-2, iNOS, and cytokines [26], and has been confirmed to reduce MFB lesions and decrease inflammatory biomarkers such as TNF-α and IL-1β [30]. Furthermore, EA can reduce the activation of NLRP3 signaling associated with neuroinflammation [30] and the secretion of pro-inflammatory factors in microglia and has a neuroprotective effect against LPS-induced dopamine neuronal damage as well [31].
PD threatens our society with a substantial health and economic burden. To slow down or halt disease progression, developing novel therapeutic strategies acting on the underlying disease pathogenesis has become an urgent necessity. Based on the effects of EA on oxidative stress and chronic inflammation in the progress of PD, EA may be a potential drug in clinical therapy.
3. EA in Cerebral Ischemia
Cerebral ischemia/reperfusion is a kind of serious cerebrovascular disease with a high morbidity and fatality rate. This ischemia can induce hypoxia in brain tissue and lead to a series of oxidative stress and inflammatory responses, ultimate dysfunction, and even death of the neuron [32].
When cerebral ischemia happens, some enzymes such as succinate dehydrogenase and NADPH oxidase in the cellular mitochondria produce more ROS. The accumulation of ROS could directly injure the intracellular structure and induce neuronal apoptosis [33]. On the other hand, cerebral ischemia induces NOS to produce NO, and excess NO can react with the superoxide anion to produce peroxynitrite and interfere with the SOD activity [34].
The accumulation of inflammatory cells and cytokines is another feature of cerebral ischemia. Exogenous mononuclear phagocytes, T lymphocytes, and polymorphonuclear leukocytes can all trigger an inflammatory response in CNS [35]. If the inflammatory response lasts for a long period of time, it can induce neuroglial cell proliferation and brain atrophy [35]. It has been seen that IL-6 and TNF-α increase significantly in cerebral ischemia and inhibiting the IL-6 and TNF-α pathways can alleviate cerebral ischemia injury [36]. The increased expression of NLRP1 and NLRP3 is also related to cerebral ischemia injury [37][38]. Researchers have shown that NLRP1 and NLRP3 contribute to energy depletion, acidosis, tissue protease release, and increased ROS production [38]. However, the mechanisms of the activation of NLRP1 and NLRP3 receptors during cerebral ischemia remain unknown.
EA can scavenge free radicals generated during cerebral ischemia, increase the activity of antioxidant enzymes, and inhibit the expression of inflammatory factors such as IL-1β and TNF-α by activating the MAPK and Nf-κB pathways to reduce cell death [39]. In addition, the other roles of EA in cerebral ischemia include: (i) improving the decreased blood-brain barrier (BBB) permeability after cerebral ischemia by inhibiting the release of inflammatory cytokines [39][40] and (ii) increasing the activity of lactate dehydrogenase and reducing the damage of lactic acid accumulation to cells [40].
EA has protective effects on the ischemic brain, and as an antioxidant and anti-inflammatory natural substance, the side effects are low. However, the exact mechanism of its action still needs further investigation.
References
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89.
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alz-heimer’s disease. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2000, 1502, 139–144.
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med Chem. 2005, 12, 1161–1208.
- Markesbery, W.R. The Role of Oxidative Stress in Alzheimer Disease. Arch. Neurol. 1999, 56, 1449–1452.
- Muthaiyah, B.; Essa, M.M.; Chauhan, V.; Chauhan, A. Protective Effects of Walnut Extract Against Amyloid Beta Peptide-Induced Cell Death and Oxidative Stress in PC12 Cells. Neurochem. Res. 2011, 36, 2096–2103.
- Javaid, N.; Shah, M.A.; Rasul, A.; Chauhdary, Z.; Saleem, U.; Khan, H.; Ahmed, N.; Uddin, M.S.; Mathew, B.; Behl, T.; et al. Neuroprotective Effects of Ellagic Acid in Alzheimer’s Disease: Focus on Underlying Mo-lecular Mechanisms of Therapeutic Potential. Curr. Pharm. Des. 2021, 27, 3591–3601.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Lin, L.; Zheng, L.J.; Zhang, L.J. Neuroinflammation, Gut Microbiome, and Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8243–8250.
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory ef-fects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689.
- Chaney, A.; Williams, S.R.; Boutin, H. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J. Neurochem. 2019, 149, 438–451.
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimers Dis. 2016, 54, 853–857.
- Goudarzi, M.; Amiri, S.; Nesari, A.; Hosseinzadeh, A.; Mansouri, E.; Mehrzadi, S. The possible neuroprotective effect of ellagic acid on sodium arsenate-induced neurotoxicity in rats. Life Sci. 2018, 198, 38–45.
- Sanadgol, N.; Golab, F.; Mostafaie, A.; Mehdizadeh, M.; Abdollahi, M.; Sharifzadeh, M.; Ravan, H. Ellagic acid ameliorates cuprizone-induced acute CNS inflammation via restriction of microgliosis and down-regulation of CCL2 and CCL3 pro-inflammatory chemokines. Cell. Mol. Biol. 2016, 62, 24–30.
- Jha, A.B.; Panchal, S.S.; Shah, A. Ellagic acid: Insights into its neuroprotective and cognitive enhancement effects in sporadic Alzheimer’s disease. Pharmacol. Biochem. Behav. 2018, 175, 33–46.
- Kiasalari, Z.; Heydarifard, R.; Khalili, M.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Zahedi, E.; Sanaierad, A.; Roghani, M. Ellagic acid ameliorates learning and memory deficits in a rat model of Alz-heimer’s disease: An exploration of underlying mechanisms. Psychopharmacology 2017, 234, 1841–1852.
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Appiani, M.C.; de Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011.
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90.
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91.
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491.
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031.
- Noyce, A.; Msc, J.P.B.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901.
- Pajares, M.; I Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687.
- Raichur, A.; Vali, S.; Gorin, F. Dynamic modeling of alpha-synuclein aggregation for the sporadic and genetic forms of Parkin-son’s disease. Neuroscience 2006, 142, 859–870.
- Zhang, F.; Liu, J.; Shi, J. Anti-inflammatory activities of resveratrol in the brain: Role of resveratrol in microglial activation. Eur. J. Pharmacol. 2010, 636, 1–7.
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909.
- Ardah, M.; Bharathan, G.; Kitada, T.; Haque, M. Ellagic Acid Prevents Dopamine Neuron Degeneration from Oxidative Stress and Neuroinflammation in MPTP Model of Parkinson’s Disease. Biomolecules 2020, 10, 1519.
- Baluchnejadmojarad, T.; Rabiee, N.; Zabihnejad, S.; Roghani, M. Ellagic acid exerts protective effect in intrastriatal 6-hydroxydopamine rat model of Parkinson’s disease: Possible involvement of ERβ/Nrf2/HO-1 signaling. Brain Res. 2017, 1662, 23–30.
- Sarkaki, A.; Farbood, Y.; Dolatshahi, M.; Mansouri, S.M.T.; Khodadadi, A. Neuroprotective Effects of Ellagic Acid in a Rat Model of Parkinson’s Disease. Acta Med. Iran. 2016, 54, 494–502.
- Tancheva, L.P.; Lazarova, M.I.; Alexandrova, A.V.; Dragomanova, S.T.; Nicoletti, F.; Tzvetanova, E.R.; Hodzhev, Y.K.; Kalfin, R.E.; Miteva, S.A.; Mazzon, E.; et al. Neuroprotective Mechanisms of Three Natural Antioxidants on a Rat Model of Parkinson’s Disease: A Comparative Study. Antioxidants 2020, 9, 49.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678.
- He, X.M.; Zhou, Y.Z.; Sheng, S.; Li, J.J.; Wang, G.Q.; Zhang, F. Ellagic Acid Protects Dopamine Neurons via Inhibition of NLRP3 In-flammasome Activation in Microglia. Oxid. Med. Cell. Longev. 2020, 2020, 2963540.
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198.
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochon-drial ROS. Nature 2014, 515, 431–435.
- Rodrigo, R.; Fernández-Gajardo, R.; Gutiérrez, R.; Manuel Matamala, J.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeu-tic opportunities. CNS Neurol. Disord. Drug Targets. 2013, 12, 698–714.
- Brea, D.; Sobrino, T.; Ramos-Cabrer, P.; Castillo, J. Inflammatory and neuroimmunomodulatory changes in acute cerebral ische-mia. Cerebrovasc. Dis. 2009, 27 (Suppl. 1), 48–64.
- Xing, J.; Lu, J. HIF-1α Activation Attenuates IL-6 and TNF-α Pathways in Hippocampus of Rats Following Transient Global Ischemia. Cell. Physiol. Biochem. 2016, 39, 511–520.
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375.
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of in-flammasomes. Ageing Res. Rev. 2013, 12, 941–966.
- Hassonizadeh Falahieh, K.; Sarkaki, A.; Edalatmanesh, M.; Gharib Naseri, M.K.; Farbood, Y. Ellagic acid attenuates post-cerebral ischemia and reperfusion behavioral deficits by decreasing brain tissue inflammation in rats. Iran. J. Basic Med. Sci. 2020, 23, 645–653.
- Pang, X.; Li, T.; Feng, L.; Zhao, J.; Zhang, X.; Liu, J. Ellagic acid-induced thrombotic focal cerebral ischemic model in rats. J. Pharmacol. Toxicol. Methods 2014, 69, 217–222.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
26 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No