 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Caterina Peggion | + 2916 word(s) | 2916 | 2020-11-11 06:41:01 | | | |
| 2 | Bruce Ren | Meta information modification | 2916 | 2020-11-18 04:31:28 | | | | |
| 3 | Maria Lina Massimino | + 3 word(s) | 2919 | 2020-11-20 10:01:03 | | |
Video Upload Options
Prion diseases are rare transmissible neurodegenerative disorders caused by the accumulation of a misfolded isoform (PrPSc) of the cellular prion protein (PrPC) in the central nervous system (CNS). Neuropathological hallmarks of prion diseases are neuronal loss, astrogliosis, and enhanced microglial proliferation and activation. As immune cells of the CNS, microglia participate both in the maintenance of the normal brain physiology and in driving the neuroinflammatory response to acute or chronic (e.g., neurodegenerative disorders) insults. Microglia involvement in prion diseases, however, is far from being clearly understood.
1. Introduction–The Prion Protein, Prions and Prion Diseases
The cellular prion protein, PrPC, is a glycosylphosphatidylinositol (GPI)-anchored protein, residing in the outer leaflet of the plasma membrane (PM), expressed in several cell types and particularly abundant in the central nervous system (CNS) and immune cells [1].
PrPC is primarily renowned for being the precursor of prions, the proteinaceous infectious agents lacking nucleic acids that cause invariably fatal neurodegenerative disorders named transmissible spongiform encephalopathies (TSEs) or prion diseases in humans and other mammalian species [2][3][4]. Prion diseases are rare disorders, affecting about one or two people per million per year world-wide, nevertheless they attract remarkable attention due to the unique biology of the transmissible agent. Prions form upon the conversion of PrPC into an insoluble, protease-resistant conformer (PrPSc) that is the major, if not unique, component of the infectious particle. Although all TSEs share the presence of PrPSc aggregate deposition, they present with a variety of etiological and neuropathological traits, affecting different CNS areas, and causing various clinical manifestations.
The great majority of human prion diseases arise sporadically (e.g., sporadic Creutzfeldt–Jakob disease (CJD)), but a significant percentage (~10%) is of genetic origin, due to the autosomal dominant transmission of inherited mutations in the PrPC-coding gene (e.g., genetic CJD, Gerstmann–Sträussler–Scheinker syndrome and fatal familial insomnia) and about 5% of all cases develop on infectious grounds (e.g., Kuru, iatrogenic CJD and a new variant form of CJD that was transmitted to humans through the consumption of meat from bovine spongiform encephalopathy-affected cattle) [5].
The classical diagnostic triad of TSEs comprises spongiform vacuolation of the cerebral grey matter, neuronal loss, and chronic neuroinflammation [6]. Regarding the latter aspect, activation and proliferation of microglia and astrocytes have been recognized as obligatory features of the disease, regardless of the TSE form (reviewed in [7][8][9]). However, the determination of the precise role of glia in prion diseases and other neurodegenerative diseases, which are commonly associated with chronic neuroinflammation, is still a matter of debate [10][11][12][13][14]. Taking this context, it is worth remarking that the cellular mechanisms leading to brain damage in TSEs are far from being understood, and it is not yet clear if they rely on a gain of toxicity of PrPSc aggregates or a loss of function of PrPC (due to its continuous conversion into PrPSc), or both. Concerning the loss of function hypothesis, the understanding of PrPC physiological function would be of paramount importance. While both the high evolutionary conservation and the almost ubiquitous expression of PrPC would suggest fundamental roles for the protein, and in spite of decades of extensive research, a comprehensive view of PrPC function, however, is still missing.
The possible involvement of PrPC in microglia pathophysiology and the role of microglia in PrPSc infection and propagation during prion disease are the major topic of this review, and, particularly, we will discuss previous controversial results on the role of PrPC on microglial activation and cytokine release.
2. Microglia Function in Health and Prion Diseases
2.1. Microglia Origin and Function in the Healthy Brain.
Microglia belong to the glial system of non-neuronal cells and represent the resident immune population of the CNS. Although it has been studied for decades, the developmental origin of microglia remained debated for a long time [15]. Recent studies confirmed the prediction of the founder of the microglia field, Pio del Rio–Hortega [16], indicating that microglial cells are derived from c-Kit+ erythromyeloid precursors in the yolk sac that seed the CNS rudiment from the cephalic mesenchyme very early during embryogenesis and continuing until the blood–brain barrier is formed, following a gradual process of differentiation into highly specialized immune cells in the brain [17][18]. After such a developmental origin, self-renewal is the only source of new microglial cells in the healthy brain, which is regulated also by astrocytes and neurons through the activation of the microglial colony-stimulating factor (CSF)-1 tyrosine kinase receptor (CSF-1R) by its ligands CSF-1 and interleukin (IL)-34 [19].
Microglial cells play a key role in the maintenance of brain homeostasis from early development to adulthood, including neurodevelopment, synaptic pruning, neuronal circuit maturation [20][21], and the impairment of microglial functions which can lead to severe pathological outcomes.
Under healthy conditions, microglial cells are characterized by a small cell soma and numerous branching processes involved in the clearance of metabolic products and apoptotic cells or cell debris [22]. Key surface receptors (i.e., cluster of differentiation (CD) 45, CD14, and CD11b/CD18 (Macrophage-1 antigen)) maintain microglial cells in a resting but highly dynamic state [23] that is favored by the interaction with neurons, for example through the formation of a molecular complex between the neuronal transmembrane glycoprotein CD200 and its receptor CD200R present in the plasma membrane of microglial cells [24], or between the neuronal chemokine C-X3-C motif chemokine (CX3C) ligand 1 (CX3CL1, also named Fractalkine) and its receptor CX3CR1, expressed solely by microglial cells [25].
2.2. Microglia Involvement in Neuropathology.
Under pathological circumstances, such as brain injury, microbial infection, or neurodegeneration, microglial activation leads to morphological changes, up-regulation of surface receptors, secretion of a multitude of cytokines, chemokines, and reactive oxygen species (ROS) [26], and the acquisition of a phagocytic phenotype [27]. Most in vitro and in vivo studies used the bacterial cell wall endotoxin lipopolysaccharide (LPS) (reviewed in [28][29][30]) or pro-inflammatory cytokines (e.g., interferon-γ (IFNγ) and tumor necrosis factor-α (TNF-α)) to achieve microglial activation [31].
Activated microglia could exist in a range of activation states that span from two opposite phenotypes, the pro-inflammatory (M1) and the anti-inflammatory (M2) phenotype, with the phenotype of activated microglia falling somewhere along this spectrum, depending on incoming signals. A precise balance between M1 and M2 is of paramount importance for the resolution of inflammation. Indeed, the M1 phase is characterized by the secretion of pro-inflammatory mediators, such as TNF-α, IL-1β, IL-6, nitric oxide (NO), ROS, chemokines (e.g., macrophage inflammatory protein 1α, monocyte chemoattractant protein-1 (MCP-1) and the IFN inducible protein 10) and neurotoxins. When such a strong pro-inflammatory response protracts over time (e.g., under chronic conditions such as neurodegenerative disorders), the sustained release of inflammatory mediators and oxidative and nitrosative stress may exacerbate neuronal damage [32][33], a circumstance that may well occur in prion diseases (see below, [9]). Conversely, in the M2 response, microglia exert numerous beneficial effects through the release of anti-inflammatory factors (e.g., IL-4, IL-10, transforming growth factor-β (TGF-β), insulin-like growth factor 1 and brain-derived neurotrophic factor) [34][35], protecting against brain damage.
2.3. Microglial Proliferation/Activation in Prion Diseases.
An active involvement of microglial activation and cytokine signaling in prion diseases has been suggested by a wealth of observations reporting different microglial responses in human CJD [36][37], in bovine spongiform encephalopathies [38], in scrapie [39], in animal models of the diseases (i.e., rodents [40][41][42] or sheep [43] infected with experimental prion strains, or prion mimetics, see below) and in cultured microglial cells exposed to PrPSc [44].
Both in patient and in animal models of prion diseases, PrPSc aggregates have been found inside and around microglial cells, inducing activation that correlates temporally with the onset and development of clinical and molecular signs of the disease [45][46][47][48][49][50][51][52][53].
The huge experimental effort, however, failed to precisely define the microglial role in prion disease progression (for a comprehensive review, see [7]). It is well-accepted, on one hand, that microglial activation begins in the early stages of disease, preceding neuronal loss and spongiform neurodegeneration, which would suggest that microglial activation is a cause rather than a consequence of neuronal demise (reviewed in [9]). A wealth of evidence, on the other hand, supports the hypothesis that microglia play a neuroprotective role in prion disease-related neuronal damage [7].
One of the most exploited models for studying prion diseases is that involving the use of mice infected with different prion strains, which recapitulated most pathological features of TSEs. Using this experimental setting, the involvement of microglia in prion diseases was principally investigated in mice in which microglial activation and cytokine signalling was altered either genetically or pharmacologically [54][55][56].
Probably due to the variety of the experimental paradigms, however, such approaches provided conflicting conclusions on the role of microglia in prion disease pathogenesis, as discussed below.
Some studies showed a beneficial role of microglia activation in prion diseases. They demonstrated, for example, that Toll-like receptors (TLRs) are involved in prion-induced microglial activation, exploiting a protective function during disease pathogenesis and suggesting that TLR signalling controls the progression of prion disease, as also indicated by the finding that a loss-of-function mutation in TLR4 decreased survival after prion infection [57].
Conversely, however, it also has been suggested that the chronic activation of microglia might have a detrimental effect in the defence against prions. Particularly, a key role in the sustained maintenance of activated microglia in prion disease is retained by the CSF-1R signalling pathway. Indeed, a reduction of proliferating microglia was observed in PrPSc-infected mice treated with the selective inhibitor of CSF-1R, GW2580, with a consequent slowdown in neuronal damage and disease progression [56].
Furthermore, the expression of the microglial GPI-anchored protein CD14, a TLR co-receptor involved in microglia activation, significantly increases in mice infected with different prion strains, as demonstrated by large-scale transcriptomic studies [58][59]. Interestingly, PrPSc-infected CD14 knock-out (KO) mice survived longer and expressed more anti-inflammatory cytokines (such as IL-10 and TGF-β [54], IL-13 [60]) and less pro-inflammatory IL-1β than PrPSc-infected wild-type (WT) mice [54], suggesting a harmful role for CD14-mediated signalling in prion pathogenesis (Figure 1).
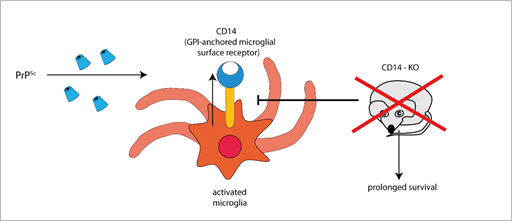
Figure 1. The GPI-anchored cell surface TLR co-receptor CD14 is upregulated in microglial cells during prion infection, while CD14 genetic deletion blocks (T-arrow) CD14 upregulation and prolongs survival in prion infected knock-out mice (red × symbol).
Another key issue is the involvement of astroglial cells in prion replication, and propagation to neighboring cells (i.e., neurons) and throughout the CNS, which has been proposed to occur in prion diseases [61] and other neurodegenerative disorders associated with prion-like proteins [62]. While the direct participation of astrocytes and neurons in PrPSc replication and spread is well recognized [63][64][65][66][67][68][69], the role of microglia in such processes is still debated. The low basal levels of PrPC in microglia may suggest that these cells unlikely support the PrPC-to-PrPSc transition and act as foci of prion propagation. Nonetheless, the increase of PrPC levels in microglia under inflammatory conditions, such as those occurring during prion infection or—experimentally—upon microglia activation by LPS, may enhance the capability of microglial cells to favor prion replication and spreading [70].
Taking this context, it also is worth noting that in other neurodegenerative proteinopathies, sharing with prion diseases the molecular mechanisms of propagation of aberrant protein conformers [71][72], the contribution of microglia in cell-to-cell transmission of proteinaceous neuropathogens has been proposed. As examples, this is the case for α-synuclein in Parkinson’s disease models [73], and of the tau protein in Alzheimer’s disease-related tauopathies [74][75][76].
2.4. Cytokines/Chemokines Signalling Alteration in Prion Diseases.
Another important and widely studied issue related to microglial response in prion diseases concerns the role of cytokines and chemokines. As previously described, both pro- and anti-inflammatory cytokines are increased in the CNS in response to prion infection. These molecules are produced by activated microglia and astrocytes, since leukocytes from the periphery do not infiltrate the CNS in prion diseases, and exert different functions in the inflammatory response to brain injury, as described above.
Regarding whole brain or isolated microglial cells of PrPSc-infected mice, an upregulation of anti-inflammatory cytokines and chemokines (principally in the early stages of prion infection) [77], or of pro-inflammatory ones [78][79][80][81][82][83][84][85][86][87] have been observed. The different results obtained in such studies may be attributable to several factors, including the used prion strains, the stage of disease, different mouse genetic backgrounds, or techniques applied for the analyte detection.
Among the pro-inflammatory cytokines, IL-1 seems to play a prominent detrimental role in prion-associated neuroinflammation, since knocking-out the IL-1 receptor 1 (IL-1R1) prolongs prion incubation times and delays disease progression in scrapie infected IL-1R1 KO [88][89].
The role of anti-inflammatory cytokines in prion diseases has been mainly investigated by means of IL-4, IL-10, and IL-13 KO mice. Among these cytokines, the only one playing a protective role in prion diseases is IL-10, as suggested by the finding that prion-inoculated IL-10 KO mice have a faster onset and progression of the disease. IL-10 absence also favors TNF-α expression, thereby promoting a sustained pro-inflammatory response [90]. However, conflicting results were obtained in IL-10 KO mice with a different genetic background [89], supporting the idea that the IL-10-neuroprotective effect is strongly dependent on the genetic context (Figure 2).
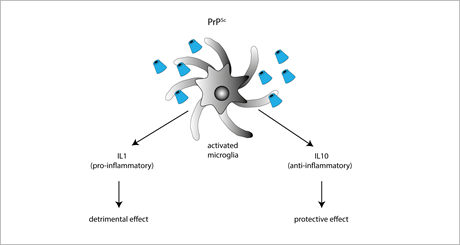
Figure 2. Exposure to PrPSc or prion mimetics (e.g., the fibrillogenic 106-126 peptide) activates microglial cells promoting the secretion of either pro-inflammatory IL-1 (resulting in enhanced neurodegenerative cues), or anti-inflammatory IL-10 (exerting neuroprotective effects).
Also, numerous chemokines (e.g., MCP-1, Regulated upon Activation, Normal T cell Expressed, and Secreted (RANTES), C-X-C motif ligand (CXCL)3, CXCL9, CXCL10) and chemokine receptors (CCR) (e.g., CCR1, CCR5 and CCR3) are up-regulated in prion diseases, but their role in the pathology remains unclear since different studies provided conflicting results. Signalling pathways activated by CXCL9 and CXCL10 seem to directly contribute to prion disease progression, as suggested by the finding that scrapie infected-CXCR3 KO mice (lacking the CXCL9 and CXCL10 receptor), although exhibiting exacerbated astrocytosis and accelerated accumulation of PrPSc, showed reduced microglia activation and pro-inflammatory factor secretions, and survived longer compared to scrapie-infected WT mice [91]. In contrast, prion infected-CCR1 KO mice (lacking the RANTES receptor) showed a worsened prion disease course and a lower survival rate with respect to infected WT mice [92].
Considering this, animal models provided conflicting results also for the involvement of the CX3CL1/CX3CR1 signalling axis in prion neuropathology. While Grizenkova et al., suggested that CX3CR1 (a receptor for the CX3CL1 chemokine) activation is neuroprotective, a subsequent study demonstrated no contribution of the receptor to disease progression [93].
2.5. Microglial Activation by the Prion Mimetic PrP106-126 Neurotoxic Peptide.
In light of the above reported controversial results provided by animal models, it is worth discussing a simpler experimental model that has been widely employed to study the microglia contribution to prion-related neurodegenerative processes. We specifically refer to the use of the fibrillogenic and neurotoxic peptide 106-126 derived from the human prion protein sequence (PrP106-126)[94], characterized by its intrinsic capability to form fibrils in vitro [95] and to induce a PrP-dependent neuronal cell death [96] by the activation of apoptotic cell death pathways [97]. Interestingly, the PrP106-126 neurotoxic effect requires the presence of microglia [98] and was demonstrated to be independent from de novo generation of PrPSc [99].
Particularly the PrP106-126 peptide, or other longer peptides containing the PrP106-126 sequence, induced the activation and proliferation of immortalized microglial cells [100][101][102] and primary microglia cultures [103][104][105][106][107][108][109][110][111], and stimulated astroglial proliferation [112][113]. Furthermore, such fibrillogenic peptides induced the secretion of different pro-inflammatory cytokines (i.e., TNF-α, [105]; RANTES, Granulocyte-CSF, and IL-12, [100]; IL-1β, IL-6, [104][106]), and caused an increase of NO synthase and NO release that appears to be essential for the mediation of neurotoxicity [105], as demonstrated by the neuronal death induced in neurons co-cultured with microglia treated with the PrP106-126 peptide [106] (Figure 2).
Taken together, the above summarized findings—based on the use of PrP-derived peptides—support the contention that microglial cells contribute to prion-related neurodegenerative processes by producing pro-inflammatory cytokines and oxidative stress, which are recognized as mediators of neuronal death. Nonetheless, it is worth underlining that some doubts on the relevance and validity of results obtained with prion protein-derived peptides were forwarded, since such studies often disregarded the non-infectious nature of PrP106-126, its absence in clinical cases, or its spontaneous production in in vivo experimental settings.
3. Concluding remarks.
As described in this review, microglia are likely to play a relevant role in prion neuropathology, but – in light of the conflicting data accumulated over time – it is far to be clearly understood if microglia play as a beneficial actor in, or an enhancer of, prion-related neurodegeneration.
One of the most accredited pictures emerging from the above discussed reports envisages that – in the first stages of the disease – microglial cells act as a suppressor of neurotoxicity induced by PrPSc deposits by facilitating their removal and clearance and by secreting anti-inflammatory factors that prevent neuronal loss.
However, in an advanced stage of disease, microglia would no longer be able to contrast the effects of prolonged PrPSc accumulation and neurotoxic signalling, which would rather elicit the switching of microglia to a sustained/chronic pro-inflammatory response, thus worsening brain damage (see following scheme).
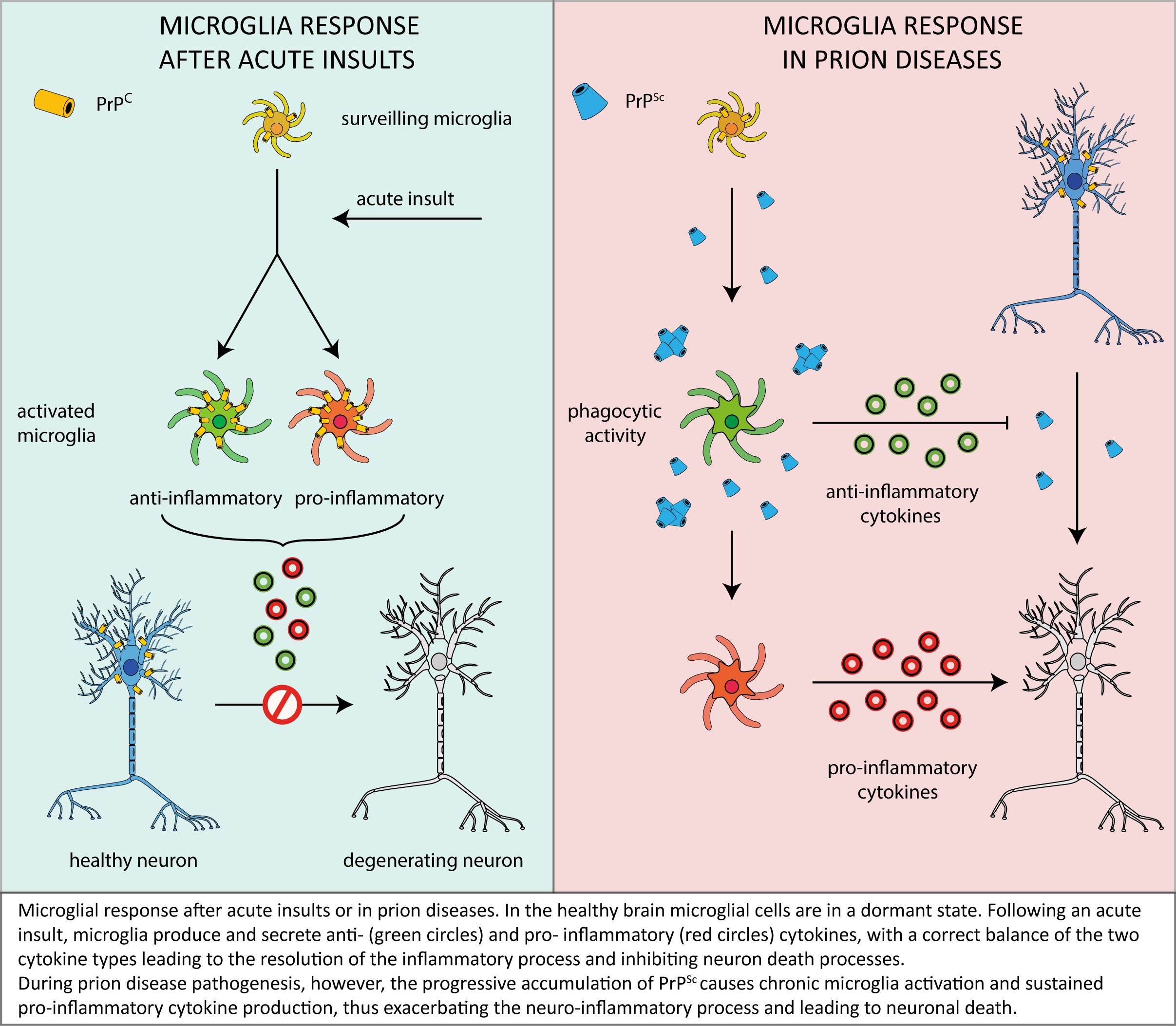
In this context, it is also worth considering that the depletion of functional PrPC in microglia and microglia-neuron crosstalk may exacerbate the effects of microglia misregulation, thereby contributing to disease progression. Also in this case, however, past literature offer a panel of conflicting data, thus highlighting once more the need for a deeper understanding of the physiological PrPC function in different cellular environments and signalling pathways.
It is our opinion that a unifying definition of microglia role in prion disease pathogenesis is crucial for both getting major insights into the neurodegenerative processes and developing suited therapeutic strategies. In particular, a prompt stimulation of the anti-inflammatory properties of microglia in the first stages of prion diseases may represent an optimal target to ameliorate disease progression and the quality of life of patients.
References
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19.
- Prusiner, S.B. Prions. Sci. Am. 1984, 251, 50–59.
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383.
- Prusiner, S.B. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513.
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559.
- DeArmond, S.J.; Prusiner, S.B. Etiology and pathogenesis of prion diseases. Am. J. Pathol. 1995, 146, 785–811.
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Investig. 2017, 127, 3230–3239.
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia Are Critical in Host Defense against Prion Disease. J. Virol. 2018, 92, e00549-18.
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207.
- Perry, V.H.; Cunningham, C.; Boche, D. Atypical inflammation in the central nervous system in prion disease. Curr. Opin. Neurol. 2002, 15, 349–354.
- Schwartz, M.; Baruch, K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014, 33, 7–22.
- Gomez-Nicola, D.; Perry, V.H. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neuroscientist 2015, 21, 169–184.
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621.
- Streit, W.J.; Xue, Q.S. Human CNS immune senescence and neurodegeneration. Curr. Opin. Immunol. 2014, 29, 93–96.
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899.
- Rio-Hortega, P. The microglia. Lancet 1939, 233, 1023–1026.
- Ginhoux, F.; Prinz, M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537.
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada González, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670.
- Chitu, V.; Gokhan, Ş.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393.
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458.
- Ikegami, A.; Haruwaka, K.; Wake, H. Microglia: Lifelong modulator of neural circuits. Neuropathology 2019, 39, 173–180.
- Raivich, G. Like cops on the beat: The active role of resting microglia. Trends Neurosci. 2005, 28, 571–573.
- Korzhevskii, D.E.; Kirik, O. V Brain Microglia and Microglial Markers. Neurosci. Behav. Physiol. 2016, 46, 284–290.
- Shrivastava, K.; Gonzalez, P.; Acarin, L. The immune inhibitory complex CD200/CD200R is developmentally regulated in the mouse brain. J. Comp. Neurol. 2012, 520, 2657–2675.
- Wolf, Y.; Yona, S.; Kim, K.W.; Jung, S. Microglia, seen from the CX3CR1 angle Front. Cell. Neurosci. 2013, 7, 26.
- Kim, S.U.; de Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313.
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318.
- Lund, S.; Christensen, K.V.; Hedtjärn, M.; Mortensen, A.L.; Hagberg, H.; Falsig, J.; Hasseldam, H.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J. Neuroimmunol. 2006, 180, 71–87.
- Andersson, P.B.; Perry, V.H.; Gordon†, S. The acute inflammatory response to lipopolysaccharide in cns parenchyma differs from that in other body tissues. Neuroscience 1992, 48, 169–186.
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114.
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215.
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69.
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47.
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65.
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98.
- Sasaki, A.; Hirato, J.; Nakazato, Y. Immunohistochemical study of microglia in the Creutzfeldt-Jakob diseased brain. Acta Neuropathol. 1993, 86, 337–344.
- Mühleisen, H.; Gehrmann, J.; Meyermann, R. Reactive microglia in Creutzfeldt-Jakob disease. Neuropathol. Appl. Neurobiol. 1995, 21, 505–517.
- Vidal, E.; Márquez, M.; Tortosa, R.; Costa, C.; Serafín, A.; Pumarola, M. Immunohistochemical approach to the pathogenesis of bovine spongiform encephalopathy in its early stages. J. Virol. Methods 2006, 134, 15–29.
- Vidal, E.; Acín, C.; Foradada, L.; Monzón, M.; Márquez, M.; Monleón, E.; Pumarola, M.; Badiola, J.J.; Bolea, R. Immunohistochemical Characterisation of Classical Scrapie Neuropathology in Sheep. J. Comp. Pathol. 2009, 141, 135–146.
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998, 8, 449–457.
- Baker, C.A.; Lu, Z.Y.; Zaitsev, I.; Manuelidis, L. Microglial activation varies in different models of Creutzfeldt-Jakob disease. J. Virol. 1999, 73, 5089–5097.
- Baker, C.A.; Martin, D.; Manuelidis, L. Microglia from Creutzfeldt-Jakob disease-infected brains are infectious and show specific mRNA activation profiles. J. Virol. 2002, 76, 10905–10913.
- González, L.; Martin, S.; Begara-McGorum, I.; Hunter, N.; Houston, F.; Simmons, M.; Jeffrey, M. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J. Comp. Pathol. 2002, 126, 17–29.
- Srivastava, S.; Katorcha, E.; Makarava, N.; Barrett, J.P.; Loane, D.J.; Baskakov, I. V Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci. Rep. 2018, 8, 11326.
- Miyazono, M.; Kitamoto, T.; Iwaki, T.; Tateishi, J. Colocalization of prion protein and β protein in the same amyloid plaques in patients with Gerstmann-Sträussler Syndrome. Acta Neuropathol. 1992, 83, 333–339.
- Barcikowska, M.; Liberski, P.P.; Boellaard, J.W.; Brown, P.; Gajdusek, D.C.; Budka, H. Microglia is a component of the prion protein amyloid plaque in the Gerstmann-Sträussler-Scheinker syndrome. Acta Neuropathol. 1993, 85, 623–627.
- Guiroy, D.C.; Wakayama, I.; Liberski, P.P.; Gajdusek, D.C. Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Sträußler syndrome. Acta Neuropathol. 1994, 87, 526–530.
- Van Everbroeck, B.; Dobbeleir, I.; De Waele, M.; De Leenheir, E.; Lübke, U.; Martin, J.J.; Cras, P. Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt-Jakob and Alzheimer’s disease. Acta Neuropathol. 2004, 108, 194–200.
- Williams, A.; Lucassen, P.J.; Ritchie, D.; Bruce, M. PrP deposition, microglial activation, and neuronal apoptosis in murine scrapie. Exp. Neurol. 1997, 144, 433–438.
- Puoti, G.; Giaccone, G.; Mangieri, M.; Limido, L.; Fociani, P.; Zerbi, P.; Suardi, S.; Rossi, G.; Iussich, S.; Capobianco, R.; et al. Sporadic Creutzfeldt-Jakob disease: The extent of microglia activation is dependent on the biochemical type of PrPSc. J. Neuropathol. Exp. Neurol. 2005, 64, 902–909.
- Kercher, L.; Favara, C.; Striebel, J.F.; LaCasse, R.; Chesebro, B. Prion protein expression differences in microglia and astroglia influence scrapie-induced neurodegeneration in the retina and brain of transgenic mice. J. Virol. 2007, 81, 10340–10351.
- West Greenlee, M.H.; Lind, M.; Kokemuller, R.; Mammadova, N.; Kondru, N.; Manne, S.; Smith, J.; Kanthasamy, A.; Greenlee, J. Temporal Resolution of Misfolded Prion Protein Transport, Accumulation, Glial Activation, and Neuronal Death in the Retinas of Mice Inoculated with Scrapie. Am. J. Pathol. 2016, 186, 2302–2309.
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2016, 90, 3003–3017.
- Sakai, K.; Hasebe, R.; Takahashi, Y.; Song, C.H.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Absence of CD14 Delays Progression of Prion Diseases Accompanied by Increased Microglial Activation. J. Virol. 2013, 87, 13433–13445.
- Grizenkova, J.; Akhtar, S.; Brandner, S.; Collinge, J.; Lloyd, S.E. Microglial Cx3cr1knockout reduces prion disease incubation time in mice. BMC Neurosci. 2014, 15, 44.
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of Microglial Proliferation during Chronic Neurodegeneration. J. Neurosci. 2013, 33, 2481–2493.
- Spinner, D.S.; Cho, I.S.; Park, S.Y.; Kim, J.I; Meeker, H.C.; Ye, X.; LaFauci, G.; Kerr, D.J.; Flory, M.J.; Kim, B.S.; et al. Accelerated Prion Disease Pathogenesis in Toll-Like Receptor 4 Signaling-Mutant Mice. J. Virol. 2008, 82, 10701–10708.
- Hwang, D.; Lee, I.Y.; Yoo, H.; Gehlenborg, N.; Cho, J.H.; Petritis, B.; Baxter, D.; Pitstick, R.; Young, R.; Spicer, D.; et al. A systems approach to prion disease. Mol. Syst. Biol. 2009, 5, 252.
- Xiang, W.; Windl, O.; Wünsch, G.; Dugas, M.; Kohlmann, A.; Dierkes, N.; Westner, I.M.; Kretzschmar, H.A. Identification of Differentially Expressed Genes in Scrapie-Infected Mouse Brains by Using Global Gene Expression Technology. J. Virol. 2004, 78, 11051–11060.
- Hasebe, R.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Temporary upregulation of anti-inflammatory cytokine IL-13 expression in the brains of CD14 deficient mice in the early stage of prion infection. Biochem. Biophys. Res. Commun. 2014, 454, 125–130.
- Mabbott, N.A. Immunology of Prion Protein and Prions. Prog. Mol. Biol. Transl. Sci. 2017, 150, 203–240.
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325.
- Victoria, G.S.; Arkhipenko, A.; Zhu, S.; Syan, S.; Zurzolo, C. Astrocyte-to-neuron intercellular prion transfer is mediated by cell-cell contact. Sci. Rep. 2016, 6, 20762.
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLOS Pathog. 2016, 12, e1005551.
- Sisó, S.; Chianini, F.; Eaton, S.L.; Witz, J.; Hamilton, S.; Martin, S.; Finlayson, J.; Pang, Y.; Stewart, P.; Steele, P.; et al. Disease phenotype in sheep after infection with cloned murine scrapie strains. Prion 2012, 6, 174–183.
- Hilton, K.J.; Cunningham, C.; Reynolds, R.A.; Perry, V.H. Early Hippocampal Synaptic Loss Precedes Neuronal Loss and Associates with Early Behavioural Deficits in Three Distinct Strains of Prion Disease. PLoS ONE 2013, 8, e68062.
- Cronier, S.; Laude, H.; Peyrin, J.M. Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc. Natl. Acad. Sci. USA 2004, 101, 12271–12276.
- Hannaoui, S.; Maatouk, L.; Privat, N.; Levavasseur, E.; Faucheux, B.A.; Haïk, S. Prion propagation and toxicity occur in vitro with two-phase kinetics specific to strain and neuronal type. J. Virol. 2013, 87, 2535–2548.
- Tahir, W.; Abdulrahman, B.; Abdelaziz, D.H.; Thapa, S.; Walia, R.; Schätzl, H.M. An astrocyte cell line that differentially propagates murine prions. J. Biol. Chem. 2020, 295, 11572–11583.
- Muñoz-Gutiérrez, J.F.; Schneider, D.A.; Baszler, T.V.; Greenlee, J.J.; Nicholson, E.M.; Stanton, J.B. hTERT-immortalized ovine microglia propagate natural scrapie isolates. Virus Res. 2015, 198, 35–43.
- Peggion, C.; Sorgato, M.C.; Bertoli, A. Prions and prion-like pathogens in neurodegenerative disorders. Pathogens 2014, 3, 149–163.
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180.
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34.
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593.
- Španić, E.; Langer Horvat, L.; Hof, P.R.; Šimić, G. Role of Microglial Cells in Alzheimer’s Disease Tau Propagation. Front. Aging Neurosci. 2019, 11, 271.
- Simon, E.; Obst, J.; Gomez-Nicola, D. The Evolving Dialogue of Microglia and Neurons in Alzheimer’s Disease: Microglia as Necessary Transducers of Pathology. Neuroscience 2019, 405, 24–34.
- Cunningham, C.; Boche, D.; Perry, V.H. Transforming growth factor beta1, the dominant cytokine in murine prion disease: Influence on inflammatory cytokine synthesis and alteration of vascular extracellular matrix. Neuropathol. Appl. Neurobiol. 2002, 28, 107–119.
- Campbell, I.L.; Eddleston, M.; Kemper, P.; Oldstone, M.B.; Hobbs, M. V Activation of cerebral cytokine gene expression and its correlation with onset of reactive astrocyte and acute-phase response gene expression in scrapie. J. Virol. 1994, 68, 2383–2387.
- Williams, A.E.; van Dam, A.M.; Man-A-Hing, W.K.; Berkenbosch, F.; Eikelenboom, P.; Fraser, H. Cytokines, prostaglandins and lipocortin-1 are present in the brains of scrapie-infected mice. Brain Res. 1994, 654, 200–206.
- Kordek, R.; Nerurkar, V.R.; Liberski, P.P.; Isaacson, S.; Yanagihara, R.; Gajdusek, D.C. Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9754–9758.
- Brown, A.R.; Webb, J.; Rebus, S.; Walker, R.; Williams, A.; Fazakerley, J.K. Inducible cytokine gene expression in the brain in the ME7/CV mouse model of scrapie is highly restricted, is at a strikingly low level relative to the degree of gliosis and occurs only late in disease. J. Gen. Virol. 2003, 84, 2605–2611.
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 2005, 25, 9275–9284.
- Kim, J.I.; Ju, W.K.; Choi, J.H.; Kim, J.; Choi, E.K.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Expression of cytokine genes and increased nuclear factor-kappa B activity in the brains of scrapie-infected mice. Mol. Brain Res. 1999, 73, 17–27.
- Lee, H.P.; Jun, Y.C.; Choi, J.K.; Kim, J.I.; Carp, R.I.; Kim, Y.S. The expression of RANTES and chemokine receptors in the brains of scrapie-infected mice. J. Neuroimmunol. 2005, 158, 26–33.
- Tribouillard-Tanvier, D.; Striebel, J.F.; Peterson, K.E.; Chesebro, B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J. Virol. 2009, 83, 11244–11253.
- Baker, C.A.; Manuelidis, L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679.
- Riemer, C.; Neidhold, S.; Burwinkel, M.; Schwarz, A.; Schultz, J.; Krätzschmar, J.; Mönning, U.; Baier, M. Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 2004, 323, 556–564.
- Schultz, J.; Schwarz, A.; Neidhold, S.; Burwinkel, M.; Riemer, C.; Simon, D.; Kopf, M.; Otto, M.; Baier, M. Role of interleukin-1 in prion disease-associated astrocyte activation. Am. J. Pathol. 2004, 165, 671–678.
- Tamgüney, G.; Giles, K.; Glidden, D.V.; Lessard, P.; Wille, H.; Tremblay, P.; Groth, D.F.; Yehiely, F.; Korth, C.; Moore, R.C.; et al. Genes contributing to prion pathogenesis. J. Gen. Virol. 2008, 89, 1777–1788.
- Thackray, A.M.; McKenzie, A.N.; Klein, M.A.; Lauder, A.; Bujdoso, R. Accelerated prion disease in the absence of interleukin-10. J. Virol. 2004, 78, 13697–13707.
- Riemer, C.; Schultz, J.; Burwinkel, M.; Schwarz, A.; Mok, S.W.F.; Gültner, S.; Bamme, T.; Norley, S.; van Landeghem, F.; Lu, B.; et al. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3-/- mice. J. Virol. 2008, 82, 12464–12471.
- LaCasse, R.A.; Striebel, J.F.; Favara, C.; Kercher, L.; Chesebro, B. Role of Erk1/2 activation in prion disease pathogenesis: Absence of CCR1 leads to increased Erk1/2 activation and accelerated disease progression. J. Neuroimmunol. 2008, 196, 16–26.
- Striebel, J.F.; Race, B.; Carroll, J.A.; Phillips, K.; Chesebro, B. Knockout of fractalkine receptor Cx3cr1 does not alter disease or microglial activation in prion-infected mice. J. Gen. Virol. 2016, 97, 1481–1487.
- Forloni, G.; Chiesa, R.; Bugiani, O.; Salmona, M.; Tagliavini, F. Review: PrP 106-126-25 years after. Neuropathol. Appl. Neurobiol. 2019, 45, 430–440.
- Tagliavini, F.; Prelli, F.; Verga, L.; Giaccone, G.; Sarma, R.; Gorevic, P.; Ghetti, B.; Passerini, F.; Ghibaudi, E.; Forloni, G. Synthetic peptides homologous to prion protein residues 106-147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 9678–9682.
- Brown, D.R.; Herms, J.; Kretzschmar, H.A. Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. Neuroreport 1994, 5, 2057–2060.
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546.
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347.
- Fioriti, L.; Quaglio, E.; Massignan, T.; Colombo, L.; Stewart, R.S.; Salmona, M.; Harris, D.A.; Forloni, G.; Chiesa, R. The neurotoxicity of prion protein (PrP) peptide 106-126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol. Cell. Neurosci. 2005, 28, 165–176.
- Thellung, S.; Corsaro, A.; Villa, V.; Venezia, V.; Nizzari, M.; Bisaglia, M.; Russo, C.; Schettini, G.; Aceto, A.; Florio, T. Amino-Terminally Truncated Prion Protein PrP90-231 Induces Microglial Activation in Vitro. Ann. N. Y. Acad. Sci. 2007, 1096, 258–270.
- Bai, Y.; Li, Y.; Wang, G.; Zhou, X.; Zhao, D. PrP 106-126 altered PrP mRNA gene expression in mouse microglia BV-2 cells. Virol. Sin. 2010, 25, 440–444.
- Chang, J.; Yang, L.; Kouadir, M.; Peng, Y.; Zhang, S.; Shi, F.; Zhou, X.; Yin, X.; Zhao, D. Antibody-mediated inhibition of integrin α5β1 blocks neurotoxic prion peptide PrP106-126-induced activation of BV2 microglia. J. Mol. Neurosci. 2012, 48, 248–252.
- Brown, D.R. Prion protein-overexpressing cells show altered response to a neurotoxic prion protein peptide. J. Neurosci. Res. 1998, 54, 331–340.
- Peyrin, J.M.; Lasmézas, C.I.; Haïk, S.; Tagliavini, F.; Salmona, M.; Williams, A.; Richie, D.; Deslys, J.P.; Dormont, D. Microglial cells respond to amyloidogenic PrP peptide by the production of inflammatory cytokines. Neuroreport 1999, 10, 723–729.
- Fabrizi, C.; Silei, V.; Menegazzi, M.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Suzuki, H.; Lauro, G.M. The stimulation of inducible nitric-oxide synthase by the prion protein fragment 106--126 in human microglia is tumor necrosis factor-alpha-dependent and involves p38 mitogen-activated protein kinase. J. Biol. Chem. 2001, 276, 25692–25696.
- Garção, P.; Oliveira, C.R.; Agostinho, P. Comparative study of microglia activation induced by amyloid-beta and prion peptides: Role in neurodegeneration. J. Neurosci. Res. 2006, 84, 182–193.
- Yang, L.; Zhou, X.; Yang, J.; Yin, X.; Han, L.; Zhao, D. Aspirin inhibits cytotoxicity of prion peptide PrP106-126 to neuronal cells associated with microglia activation in vitro. J. Neuroimmunol. 2008, 199, 10–17.
- Herms, J.W.; Madlung, A.; Brown, D.R.; Kretzschmar, H.A. Increase of intracellular free Ca2+ in microglia activated by prion protein fragment. Glia 1997, 21, 253–257.
- Kouadir, M.; Yang, L.; Tan, R.; Shi, F.; Lu, Y.; Zhang, S.; Yin, X.; Zhou, X.; Zhao, D. CD36 participates in PrP(106-126)-induced activation of microglia. PLoS ONE 2012, 7, e30756.
- Tu, J.; Yang, L.; Zhou, X.; Qi, K.; Wang, J.; Kouadir, M.; Xu, L.; Yin, X.; Zhao, D. PrP106-126 and Aβ 1-42 peptides induce BV-2 microglia chemotaxis and proliferation. J. Mol. Neurosci. 2014, 52, 107–116.
- Wang, J.; Zhao, D.; Pan, B.; Fu, Y.; Shi, F.; Kouadir, M.; Yang, L.; Yin, X.; Zhou, X. Toll-like receptor 2 deficiency shifts PrP106-126-induced microglial activation from a neurotoxic to a neuroprotective phenotype. J. Mol. Neurosci. 2015, 55, 880–890.
- Forloni, G.; Del Bo, R.; Angeretti, N.; Chiesa, R.; Smiroldo, S.; Doni, R.; Ghibaudi, E.; Salmona, M.; Porro, M.; Verga, L. A neurotoxic prion protein fragment induces rat astroglial proliferation and hypertrophy. Eur. J. Neurosci. 1994, 6, 1415–1422.
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. A prion protein fragment primes type 1 astrocytes to proliferation signals from microglia. Neurobiol. Dis. 1998, 4, 410–422.

