 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael N. Romanov | -- | 5062 | 2022-10-20 14:35:49 | | | |
| 2 | Michael N. Romanov | + 6 word(s) | 5068 | 2022-10-20 14:43:51 | | | | |
| 3 | Catherine Yang | -2082 word(s) | 2986 | 2022-10-21 03:23:15 | | | | |
| 4 | Catherine Yang | Meta information modification | 2986 | 2022-10-21 03:24:58 | | |
Video Upload Options
Interchromosomal rearrangements involving microchromosomes are rare events in birds. To date, they have been found mostly in Neognathae and Neoaves (e.g., Psittaciformes, Falconiformes, and Cuculiformes), although only a few orders have been analyzed. Hence, cytogenomic studies focusing on microchromosomes in species belonging to different bird orders are essential to shed more light on the avian chromosome and karyotype evolution. Relevant hypothetical Neognathae, Neoaves and other ancestral karyotypes can be reconstructed to trace these rearrangements. In a more recent study, a comparative chromosome mapping for chicken microchromosomes 10 to 28 was performed using interspecies BAC-based FISH hybridization in five species, representing four Neoaves orders (Caprimulgiformes, Piciformes, Suliformes, and Trogoniformes). These results suggest that the ancestral microchromosomal syntenies are conserved in Pteroglossus inscriptus (Piciformes), Ramphastos tucanus tucanus (Piciformes), and Trogon surrucura surrucura (Trogoniformes). On the other hand, chromosome reorganization in Phalacrocorax brasilianus (Suliformes) and Hydropsalis torquata (Caprimulgiformes) included fusions involving both macro- and microchromosomes. Fissions in macrochromosomes were observed in P. brasilianus and H. torquata. No interchromosomal rearrangement involving microchromosomes were found to be shared between avian orders where rearrangements were detected. These findings suggest that convergent evolution involving microchromosomal change is a rare event in birds and may be appropriate in cytotaxonomic inferences in orders where these rearrangements occurred.
1. Avian Karyotypes: Macro- and Microchromosomes
Birds (class Aves) are the most diverse lineage of extant tetrapod vertebrates, comprising 10,806 extant species, divided into 40 extant avian orders [1]. A comprehensive avian phylogeny was described by Prum et al. [2]. Despite the extraordinary diversity in morphology, ecology and behavior [3], a high proportion of species analyzed so far showed karyotypes composed of about 80 chromosomes, consisting of a few large macrochromosomes (~10) and numerous microchromosomes (~30) [4][5][6]. The latter are known to belie analysis by cytogenetic means [7].
The above cytogenomic structure is mostly conserved since the Archelosaur common ancestor and is thought to be a feature of non-avian dinosaurs [8]. Some exceptions to the typical avian karyotype are seen within the superorder Neoaves from the infraclass Neognathae, including the orders Falconiformes [9], Psittaciformes [10], and Ciconiiformes [11], which have reduced diploid numbers, and Piciformes having higher diploid numbers [12][13]. The decrease or increase of chromosome number can result from fusion and fission events, respectively [14]. According to Imai et al. [15] and their “minimum-interaction hypothesis”, the karyotype evolution tends to increase the diploid number and the number of acrocentric chromosomes by centric fissions, minimizing the risk of deleterious rearrangements. In addition, these authors suggested that the relative probability of reciprocal translocations (i.e., centric fusion) declines with increases in chromosome number and in nuclear volume.
A lower number of microchromosomes in avian species with reduced diploid numbers is the most prominent karyotypic difference as compared to species with greater diploid numbers [5]. Although the analyses of nucleotide substitution patterns in two representatives of the order Galliformes from the superorder Galloanserae, chicken (Gallus gallus, GGA) and turkey (Meleagris gallopavo) [16], have revealed a higher rate of sequence evolution on microchromosomes as compared to macrochromosomes, these tiny elements appear to be highly conserved syntenically and not prone to breakage [17]. On the other hand, using cross-species fluorescent in situ hybridization (FISH), chromosome fissions have been reported for almost all the avian macrochromosomes (except GGA8 and 10) [4][5][6][18], most of them in the first five autosomal pairs (GGA1–5) [5][6]. The breakpoint regions involved in these chromosomal rearrangements are usually associated with genomic features, including transposable elements and conserved noncoding elements. It has been suggested that they are reused in avian chromosome evolution [19][20]. Overall, chromosomal rearrangements play a key role in genome evolution, fertility and genetic disease and, thus, tools for the macro- and microchromosomes are essential to analyze such phenomena in birds [7].
2. Methods to Study Macro- and Microchromosomes
2.1. Diploid Number and Karyotype Description by Giemsa Staining
To determine the diploid chromosome number and chromosomal morphologies for avian species, metaphase spreads are conventionally stained (Giemsa 5%) and analyzed. Chromosomes are numbered consecutively based on their size and centromere position [21]. Examples of a detailed karyotype description are shown in Figure 1.

Figure 1. Examples of complete Giemsa-stained karyotypes [22]: neotropic cormorant (Phalacrocorax brasilianus, Suliformes) (a), scissor-tailed nightjar (Hydropsalis torquata, Caprimulgiformes) (b), lettered aracari (Pteroglossus inscriptus, Piciformes) (c), red-billed toucan (Ramphastos tucanus tucanus, Piciformes) (d), and surucua trogon (Trogon surrucura surrucura, Trogoniformes) (e).
2.2. Whole Chromosome Painting
Also, avian karyotypes have been investigated over the last decades by whole chromosome painting using different probes sets [4][5]. These analyses have been an important tool to detect chromosomal similarities and differences between species and changes in each lineage since they diverged from common ancestors, which can also be virtually reconstructed using software algorithms (e.g., [8][23][24][25][26][27]). However, this approach has been applied to less than 1% of species, and so many avian orders have no information concerning chromosomal homology based on molecular cytogenetics [5][6]. Hence, our knowledge about chromosome organization in birds remains somewhat patchy, especially because most of the studies have included only probes corresponding to ancestral macrochromosomes (homology to GGA 1–9) [4][5].
2.3. BAC-based FISH
Libraries of large-insert genomic clones, e.g., bacterial artificial chromosome (BAC) clones, are well-known tools for molecular cytogenetic analysis of avian genomes [28]. Lithgow et al. [7] reported the development of chicken microchromosomal paint pools, generation of pairs of specific microchromosome BAC clones in chicken, and tools for in-silico genomic comparison of microchromosomes. Recently, BAC probes from the genomic libraries of chicken and zebra finch (Taeniopygia guttata, Passeriformes, Neoaves) have been applied successfully for performing FISH across multiple avian species totaling chromosome maps for 36 species from 12 different orders [17][18][29][30][31][32][33][34][35]. Examples of FISH mapping are presented in Figure 2. Results from these studies suggested evolutionary stability in avian microchromosome organization, except in Falconiformes, Psittaciformes, and Cuculiformes species, in which microchromosomal fusions were found [17][18][29][30][32][33], demonstrating the usefulness of microchromosome BAC probes to provide a more extensive analysis of chromosomal evolution in birds.
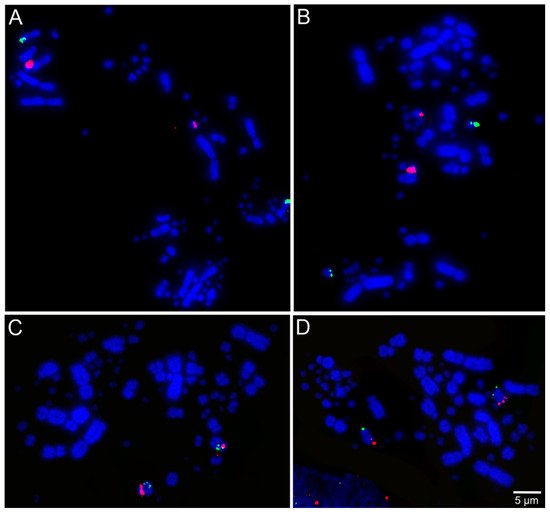
Figure 2. Examples of fluorescent in situ hybridization (FISH) experiments using chicken or zebra finch bacterial artificial chromosome (BAC) probes in the neotropic cormorant (Phalacrocorax brasilianus, Suliformes) [22]: (A) chicken macrochromosome 5 CH261-49B22 FITC and CH261-78F13 Texas red; (B) chicken macrochromosome 6 TGMCBA-382J4 FITC and CH261-49F3 Texas red; (C) chicken macrochromosome 9 CH261-183N19 FITC and chicken macrochromosome 10 CH261-115G24 Texas red; and (D) chicken microchromosome 11 CH261-154H1 FITC and chicken microchromosome 13 TGMCBA-321B13 Texas red.
3. Reconstruction of Ancestral Karyotypes
To trace chromosomal changes in the evolutionary lineages of birds, presumed Neognathae, Neoaves and other ancestral karyotypes can be reconstructed in silico. Datasets resulted from cross-species FISH-based mapping are used as input files for the software-assisted reconstruction of the hypothetical ancestral karyotypes using, for example, the maximum-likelihood algorithm. For this purpose, the Maximum Likelihood for Gene Order Analysis (MLGO) webserver [36] can be employed. The MLGO reconstruction algorithm built up a Neoaves ancestor (NAA) using the maximum-likelihood scenario for a certain number of common BACs among a selected number of avian species studied plus the chicken reference karyotype. Being quite flexible, the algorithm can handle mixed datasets with missing/failed hybridization information for chromosome location of a particular BAC in a single species. For macrochromosome datasets in chicken and other birds, information about physical positions/orders of BACs relative to each other, p/q arms and centromeres is taken into account, enabling to reconstruct ancestral macrochromosomes in more detail.
Figure 3 demonstrated an example of ancestral karyotype reconstruction [22] where the obtained interspecies FISH hybridization datasets for five Neoaves species were amended with information available from other relevant studies for six more species, including five other Neognathae/Neoaves representatives: smooth-billed ani (Crotophaga ani, Cuculiformes) [32], budgerigar (Melopsittacus undulatus, Psittaciformes) [18], saker falcon (Falco cherrug, Falconiformes) [18], peregrine falcon (Falco peregrinus, Falconiformes) [18][30], pigeon (Columba livia, Columbiformes) [27][37], and one Palaeognathae species, ostrich (Struthio camelus, Struthioniformes) [18]. This expanded dataset for 12 birds was employed for reconstructing in silico the Neognathae/Neoaves ancestral karyotypes using information about positions/orders of BACs on macrochromosomes, as well . As established in the relevant reconstruction studies [8][23][24][27], MLGO outputs were, by inference and manually, curated and adjusted further to interpret and correct software-assisted reconstruction results using the most parsimonious explanation of the available data.
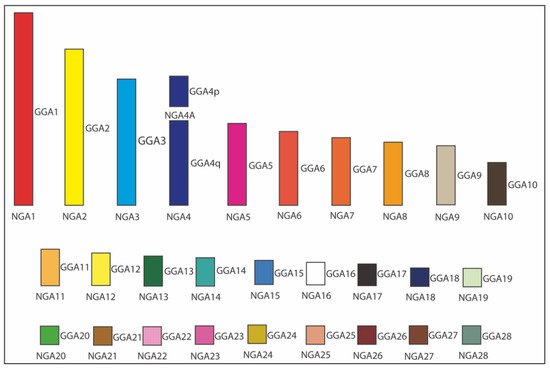
Figure 3. Ideogram of Neognathae ancestor (NGA) karyotype (NGA1 to NGA28) [22]. The NGA ancestral karyotype is likely to have the same homology with chicken (Gallus gallus; GGA), except GGA4 split into two separated chromosomes. Each GGA chromosome is illustrated with a different color. The white color indicates the probable homology with GGA16.
As can be seen in Figure 3, Kretschmer et al. [36] were able to perform an overall estimation of the hypothetical Neognathae ancestor (NGA) using the web-based MLGO tool, the relevant input files for BAC order/orientation maps in the whole set of 12 species, and the relevant input phylogenetic tree [2]. This was also possible thanks to the chromosome maps for the ostrich (infraclass Palaeognathae) available from the O’Connor et al. [18] study and used as an outgroup. In the case of macrochromosomes, the orientation of each BAC relative to its neighbors on a particular chicken chromosome and relative to p/q arms was an additional advantage for the reconstruction of bird ancestors.
As a result of the above ancestral karyotype reconstruction [22], a similar pattern of chromosome organization in the presumable NGA and Neoaves ancestor (NAA) was observed. Overall, according to this gross estimate and using datasets for the 12 species produced in this and few other published studies, NGA and NAA are likely to have 29 chromosomes (autosomes), including 10 macrochromosomes (i.e., autosomes 1–9 + 4A) and 19 microchromosomes (i.e., autosomes 10–28). Compared to the chicken karyotype, the only difference between these two karyotypes and the chicken one was that chromosome GGA4 was split into two separate chromosomes (4 and 4A) in NGA and NAA. Considering that the infraclasses Palaeognathae (ratites and tinamous) and Neognathae (superorders Galloanserae and Neoaves) diverged about 100 million years ago (Mya) and the Neognathae diverged into the evolutionary lineages of Galloanserae and Neoaves about 88 Mya [38], the most ancestral NGA karyotype can be used to compare with the FISH results.
4. Comparison of Microchromosome Organization in Birds
Kretschmer et al. [22] combined the data available in the literature about chromosomal rearrangements in 18 microchromosomes in birds. In a total of 34 avian species, nine presented interchromosomal rearrangements (Table 1). The average frequency of species with interchromosomal rearrangements along the 18 microchromosomes was 10.3%, but significant heterogeneity was found among chromosomes (χ2 = 40.927, df = 17, p = 0.001). The frequency was higher in NGA10 (26%, p = 0.001), NGA13 (21%, p = 0.042) and NGA14 (23%, p = 0.009) (Table 1). No interchromosomal rearrangements were observed in microchromosomes NGA22, NGA24, NGA26 and NGA27 (all at p < 0.05). However, when adjusting for the number of tests performed, only in NGA10 the excess of species with rearrangements reached statistical significance (p = 0.025) (Table 1).
Table 1. Number of avian species with and without interchromosomal rearrangements in 18 microchromosomes, considering 34 avian species, which had been already studied with BAC probes for microchromosomes (as summarized in [22]).
|
Microchromosome |
Interchromosomal Rearrangement |
With % |
Residual Analysis |
P Values |
Adjusted P Values |
||
|
With |
Without |
With |
Without |
||||
|
NGA10 |
9 |
25 |
26.5 |
3.19 |
−3.19 |
0.0014 |
0.0252 |
|
NGA11 |
5 |
29 |
14.7 |
0.87 |
−0.87 |
0.384 |
0.6912 |
|
NGA12 |
6 |
28 |
17.6 |
1.45 |
−1.45 |
0.147 |
0.2940 |
|
NGA13 |
7 |
27 |
20.6 |
2.03 |
−2.03 |
0.042 |
0.1080 |
|
NGA14 |
8 |
26 |
23.5 |
2.61 |
−2.61 |
0.009 |
0.0810 |
|
NGA15 |
4 |
30 |
11.8 |
0.29 |
−0.29 |
0.772 |
0.7720 |
|
NGA17 |
4 |
30 |
11.8 |
0.29 |
−0.29 |
0.772 |
0.7720 |
|
NGA18 |
3 |
31 |
8.8 |
−0.29 |
0.29 |
0.772 |
0.7720 |
|
NGA19 |
3 |
31 |
8.8 |
−0.29 |
0.29 |
0.772 |
0.7720 |
|
NGA20 |
4 |
30 |
11.8 |
0.29 |
−0.29 |
0.772 |
0.7720 |
|
NGA21 |
3 |
31 |
8.8 |
−0.29 |
0.29 |
0.772 |
0.7720 |
|
NGA22 |
0 |
34 |
0.0 |
−2.03 |
2.03 |
0.042 |
0.1080 |
|
NGA23 |
3 |
31 |
8.8 |
−0.29 |
0.29 |
0.772 |
0.7720 |
|
NGA24 |
0 |
34 |
0.0 |
−2.03 |
2.03 |
0.042 |
0.1080 |
|
NGA25 |
1 |
33 |
2.9 |
−1.45 |
1.45 |
0.147 |
0.2940 |
|
NGA26 |
0 |
34 |
0.0 |
−2.03 |
2.03 |
0.042 |
0.1080 |
|
NGA27 |
0 |
34 |
0.0 |
−2.03 |
2.03 |
0.042 |
0.1080 |
|
NGA28 |
3 |
31 |
8.8 |
−0.29 |
0.29 |
0.772 |
0.7720 |
5. Cytogenomics in Individual Neoaves Birds
In previous studies, BAC probes have been used for inter-cross FISH mapping to comprehend the structure and organization of the chromosomes in species from 12 orders, and interchromosomal rearrangements were reported only in Falconiformes, Psittaciformes and Cuculiformes species [17][18][29][30][31][32][33]. Kretschmer et al. [22] cytogenomically analyzed five species from four more Neoaves orders (Caprimulgiformes, Piciformes, Suliformes, and Trogoniformes), expanding the results to a total of 16 orders. Overall, the results demonstrated that interchromosomal rearrangements involving macro- and microchromosomes had an important role in the karyotype evolution of species of Caprimulgiformes and Suliformes, while the microchromosomes remained highly conserved in Piciformes and Trogoniformes. These results suggest that the microchromosomes NGA10, NGA13 and NGA14 are involved in multiple rearrangements. However, only in NGA10, the frequency of rearrangements was supported statistically. Karyotype information for individual Neoaves species according to [22] is provided below.
5.1. Karyotype of P. brasilianus (Suliformes)
The karyotype of P. brasilianus (Suliformes) comprises 74 chromosomes and was reported [22] for the first time. Although this diploid number is slightly lower than the “typical” avian karyotype (2n ≈ 80), Kretschmer et al. [22] detected fissions of chromosomes homologous to ancestral pairs 5 and 6 (NGA5 and NGA6). Chromosome fissions involving the homologous chromosome to NGA5 are frequent in birds and have been reported in species from the following Neoaves orders: Accipitriformes, Charadriiformes, Eurypygiformes, Falconiformes, Gruiformes, Passeriformes, Trogoniformes, Piciformes, and Strigiformes [9][35][39][40][41][42][43], while fissions in GGA6 have been detected previously only in Psittaciformes species [33][44][45] and in Crotophaga ani, a Cuculiformes species [44]. In addition, four chromosomal associations were detected in P. brasilianus, including associations between macrochromosomes (NGA5/7), macrochromosomes and microchromosomes (NGA8/12 and NGA9/10) and between microchromosomes (NGA11/13). Considering that the karyotype of P. brasilianus is similar to other Suliformes species, especially Phalacrocorax bransfieldensis, which shares the same diploid number [46], it is likely that microchromosome fusions are not exclusive to P. brasilianus in the order Suliformes.
5.2. Karyotype of H. torquata (Caprimulgiformes)
Fusion and fission events were also observed in H. torquata (Caprimulgiformes) (2n = 74). Chromosomal fissions were found in ancestral chromosome pairs 1, 2 and 5 (NGA1, 2 and 5). The breakpoints involved in these fissions are probably reused in bird chromosome evolution since fissions in these chromosomes were reported in several orders of birds [4][5][6][18]. However, the use of a higher number of BACs probes covering these chromosomes is necessary to confirm this hypothesis. In H. torquata, Kretschmer et al. [22] found fusions between macrochromosomes (NGA6/10) and between macrochromosomes and microchromosomes (NGA9/13 and NGA8/14). Based on the fact that conventional cytogenetic analyses in Caprimulgiformes species revealed an interesting range of diploid number, from 2n = 68 in Chordeiles pusillus [47] to 2n = 86 in Nyctibius griseus [48], it is plausible to infer that fusions involving microchromosomes and macrochromosomes appear to have played an important role in the chromosome evolution of this group.
5.3. Karyotype of T. s. surrucura (Trogoniformes)
Chromosomal analysis of Trogoniformes species is still rare and is based only on the karyotype description and chromosome painting in T. s. surrucura, with 2n = 82 [49]. Although a microchromosome fusion was proposed in this species based on chicken macrochromosome painting [49], Kretschmer et al. [22] did not find any evidence of this rearrangement. However, given that there are no probes available for chicken chromosomes 16 and 29–38, the occurrence of microchromosome fusions cannot be entirely discard.
5.4. Karyotype of P. inscriptus and R. t. tucanus (Piciformes)
The diploid numbers found in P. inscriptus and R. t. tucanus, 2n = 112 in both species, raise questions concerning the rearrangements that may have led to this high diploid number. Comparative chromosome painting with chicken macrochromosome probes (GGA1–10, homologous to NGA1–10) has been performed, and extensive chromosomal fissions were found in the first five ancestral chromosome pairs (NGA1–5) [13]. However, the results presented in [22] demonstrate the conservation of the ancestral patterns of microchromosomes in both species, suggesting that the fission events exclusively involved macrochromosomes. This finding suggests that the high diploid number observed in Ramphastidae species is a result of macrochromosomal fission only. Another interesting feature observed in Piciformes species is that the Z sex chromosome is the largest element of the karyotype [13][50]. Kretschmer et al. [22] excluded the possibility that a fusion event took place between the Z chromosome and any of the microchromosomes tested, corroborating a previous hypothesis that fissions of the macrochromosomes and the accumulation of repetitive sequences are the most likely mechanism responsible for the appearance of this enlarged sex chromosome in Piciformes species [13].
6. Overview of Interchromosomal Rearrangements Involving Microchromosomes in Birds
So far, no patterns of interchromosomal rearrangement have been reported as being shared among the species that exhibit rearrangements involving microchromosomes, e.g., from Cuculiformes [32], Psittaciformes [18], Falconiformes [17][30], Caprimulgiformes, and Suliformes. However, while microchromosomal fusions were shared by three Falconiformes species [17][30], each of the four Psittaciformes species studied exhibited a different pattern of microchromosomal fusions [17]. This would suggest that the convergent evolution of microchromosomal rearrangements seems to be a rare event in birds and may be an appropriate tool for phylogenetic analyses in the taxa where these rearrangements are present.
While it is evident that there are karyotypes highly rearranged and interchromosomal rearrangements involving microchromosomes were expected in Cuculiformes, Psittaciformes and Falconiformes [17][18][30][32][33], such rearrangements were not evident in the Giemsa-staining karyotypes of P. brasilianus and H. torquata, despite the fact that their diploid numbers are slightly lower than the putative avian ancestral karyotype (2n = 80) [22]. Hence, even in species with conserved karyotypes at first glance, microchromosomal fusions may have played an important role in their karyotype evolution.
Some microchromosome syntenies are involved in multiple rearrangements; for example, the frequency of species with interchromosomal rearrangements was higher in three syntenic groups, homologous to microchromosomes NGA10, NGA13 and NGA14 [22]. However, only in NGA10, the frequency of rearrangements was statistically higher than the average. Why this microchromosome is more prone to interchromosomal rearrangement than others remains unclear and deserves futures studies. Kretschmer et al. [22] did not observe rearrangements in pairs NGA22, 24, 26 and 27, but this result was not supported by the statistical analysis. A possible explanation could be the small sample size studied (34 species), but the real stability of these microchromosomes to rearrangements cannot be ruled out until new studies are done.
The above results [22] corroborate the recent suggestion that microchromosomes are not prone to breakage [17] since no fissions in these elements were observed to date. However, it is unclear why interchromosomal rearrangements involving microchromosomes are quite common in some orders (e.g., Falconiformes, Psittaciformes, Caprimulgiformes, Cuculiformes, and Suliformes), while in most other avian orders, they have remained largely unchanged [17]. Nevertheless, as more chromosome mapping of BAC probes is performed, the list of orders with microchromosome fusions is likely to increase, with some species-specific rearrangements being detected. Future cytogenomic studies using this approach will provide greater clarity on why microchromosomes remain conserved as discrete units in some species while they are prone to interchromosomal rearrangement in others.
Concluding, in this cross-species FISH mapping study, Kretschmer et al. [22] reported and characterized the organization of microchromosomes in species from four different Neoaves orders. The results have further contributed to avian cytogenomics, revealing that microchromosome fusions are not exclusive to the orders Cuculiformes, Falconiformes and Psittaciformes but are also inherent in representatives of the orders Caprimulgiformes and Suliformes. Additionally, these findings suggested that some microchromosomes are more likely to undergo interchromosomal rearrangements than others and that convergent evolution of microchromosomal rearrangements is a rare event in birds.
References
- IOC World Bird List, (v11.1); Gill, F.; Donsker, D.; Rasmussen, P. (Eds.) Gill & Wright: London, UK, 2021; Available online: https://www.worldbirdnames.org/ioc-lists/crossref/ (accessed on 22 March 2021).
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573.
- Gill, F.B. Ornithology, 2nd ed.; W.H. Freeman and Company: New York, NY, USA, 1995.
- Griffin, D.K.; Robertson, L.B.W.; Tempest, H.G.; Skinner, B.M. The evolution of the avian genome as revealed by comparative molecular cytogenetic. Cytogenet. Genome Res. 2007, 117, 64–77.
- Kretschmer, R.; Ferguson-Smith, M.A.; de Oliveira, E.H.C. Karyotype evolution in birds: From conventional staining to chromosome painting. Genes 2018, 9, 181.
- Degrandi, T.M.; Barcellos, S.A.; Costa, A.L.; Garnero, A.D.V.; Hass, I.; Gunski, R.J. Introducing the bird chromosome database: An overview of cytogenetic studies in birds. Cytogenet. Genome Res. 2020, 160, 199–205.
- Lithgow, P.E.; O’Connor, R.; Smith, D.; Fonseka, G.; Rathje, C.; Frodsham, R.; O’Brien, P.C.; Ferguson-Smith, M.A.; Skinner, B.M.; Griffin, D.K.; Romanov, M.N. Novel tools for characterising inter- and intra-chromosomal rearrangements in avian microchromosomes. In 2014 Meeting on Avian Model Systems, Cold Spring Harbor, NY, USA, 5–8 March 2014; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2014; p. 56.
- O’Connor, R.E.; Romanov, M.N.; Kiazim, L.G.; Barrett, P.M.; Farré, M.; Damas, J.; Ferguson-Smith, M.; Valenzuela, N.; Larkin, D.M.; Griffin, D.K. Reconstruction of the diapsid ancestral genome permits chromosome evolution tracing in avian and non-avian dinosaurs. Nat. Commun. 2018, 9, 1883.
- Nishida, C.; Ishijima, J.; Kosaka, A.; Tanabe, H.; Habermann, F.A.; Griffin, D.K.; Matsuda, Y. Characterization of chromosome structures of Falconinae (Falconidae, Falconiformes, Aves) by chromosome painting and delineation of chromosome rearrangements during their differentiation. Chromosome Res. 2008, 16, 171–181.
- Furo, I.O.; Kretschmer, R.; O’Brien, P.C.M.; Pereira, J.C.; Garnero, A.D.V.; Gunski, R.J.; Ferguson-Smith, M.A.; de Oliveira, E.H.C. Chromosome painting in Neotropical long- and short-tailed parrots (Aves, Psittaciformes): Phylogeny and proposal for a putative ancestral karyotype for tribe Arini. Genes 2018, 9, 491.
- Seligmann, I.C.A.; Furo, I.O.; dos Santos, M.S.; Tagliarini, M.M.; Araujo, C.C.D.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; de Oliveira, E.H.C. Comparative chromosome painting in two Brazilian stork species with different diploid numbers. Cytogenet. Genome Res. 2019, 159, 32–38.
- Castro, M.S.; Recco-Pimentel, S.M.; Rocha, G.T. Karyotypic characterization of Ramphastidae (Piciformes, Aves). Genet. Mol. Biol. 2002, 25, 147–150.
- Kretschmer, R.; Furo, I.O.; Cioffi, M.B.; Gunski, R.J.; Garnero, A.D.V.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; de Freitas, T.R.O.; de Oliveira, E.H.C. Extensive chromosomal fissions and repetitive DNA accumulation shaped the atypical karyotypes of two Ramphastidae (Aves: Piciformes) species. Biol. J. Linn. Soc. 2020, 130, 839–849.
- White, M.J.D. Animal Cytology and Evolution, 3rd ed.; Cambridge University Press: Cambridge, UK, 1973.
- Imai, H.T.; Maruyama, T.; Gojobori, T.; Inoue, Y.; Crozier, R.H. Theoretical bases for karyotype evolution. 1. The minimum-interaction hypothesis. Am. Nat. 1986, 128, 900–920.
- Axelsson, E.; Webster, M.T.; Smith, N.G.C.; Burt, D.W.; Ellegren, H. Comparison of the chicken and turkey genomes reveals a higher rate of nucleotide divergence on microchromosomes than macrochromosomes. Genome Res. 2005, 15, 120–125.
- O’Connor, R.E.; Kiazim, L.; Skinner, B.; Fonseka, G.; Joseph, S.; Jennings, R.; Larkin, D.M.; Griffin, D.K. Patterns of microchromosome organization remain highly conserved throughout avian evolution. Chromosoma 2019, 128, 21–29.
- O’Connor, R.E.; Farré, M.; Joseph, S.; Damas, J.; Kiazim, L.; Jennings, R.; Bennett, S.; Slack, E.A.; Allanson, E.; Larkin, D.M.; et al. Chromosome-level assembly reveals extensive rearrangement in saker falcon and budgerigar, but not ostrich, genomes. Genome Biol. 2018, 19, 171.
- Skinner, B.M.; Griffin, D.K. Intrachromosomal rearrangements in avian genome evolution: Evidence for regions prone to breakpoints. Heredity 2012, 108, 37–41.
- Farré, M.; Narayan, J.; Slavov, G.T.; Damas, J.; Auvil, L.; Li, C.; Jarvis, E.D.; Burt, D.W.; Griffin, D.K.; Larkin, D.M. Novel insights into chromosome evolution in birds, archosaurs, and reptiles. Genome Biol. Evol. 2016, 8, 2442–2451.
- Guerra, M.S. Reviewing the chromosome nomenclature of Levan et al. Braz. J. Genet. 1986, 9, 741–743.
- Kretschmer, R.; de Souza, M.S.; Furo, I.d.O.; Romanov, M.N.; Gunski, R.J.; Garnero, A.d.V.; de Freitas, T.R.O.; de Oliveira, E.H.C.; O’Connor, R.E.; Griffin, D.K. Interspecies chromosome mapping in Caprimulgiformes, Piciformes, Suliformes, and Trogoniformes (Aves): Cytogenomic insight into microchromosome organization and karyotype evolution in birds. Cells 2021, 10, 826. https://doi.org/10.3390/cells10040826
- Romanov, M.N.; Farré-Belmonte, M.; Lithgow, P.E.; O’Connor, R.; Fowler, K.E.; Larkin, D.M.; Griffin, D.K. In silico reconstruction of chromosomal rearrangements and an avian ancestral karyotype. In Proceedings of the International Plant and Animal Genome XXII Conference, San Diego, CA, USA, 11–15 January 2014; Scherago International: San Diego, CA, USA, 2014. Abstract P1106.
- Romanov, M.N.; Farré, M.; Lithgow, P.E.; Fowler, K.E.; Skinner, B.M.; O’Connor, R.; Fonseka, G.; Backström, N.; Matsuda, Y.; Nishida, C.; et al. Reconstruction of gross avian genome structure, organization and evolution suggests that the chicken lineage most closely resembles the dinosaur avian ancestor. BMC Genom. 2014, 15, 1060.
- Romanov, M.N.; Farré, M.; Lithgow, P.E.; O’Connor, R.E.; Fowler, K.E.; Skinner, B.M.; Larkin, D.M.; Griffin, D.K. Avian ancestral karyotype reconstruction and differential rates of inter- and intrachromosomal change in different lineages. Chromosome Res. 2015, 23, 414.
- O’Connor, R.E.; Romanov, M.N.; Farré, M.; Larkin, D.M.; Griffin, D.K. Reconstruction of the putative Saurian karyotype and the hypothetical chromosome rearrangements that occurred along the Dinosaur lineage. Chromosome Res. 2015, 23, 379–380.
- Kiazim, L.G.; O’Connor, R.E.; Larkin, D.M.; Romanov, M.N.; Narushin, V.G.; Brazhnik, E.A.; Griffin, D.K. Comparative mapping of the macrochromosomes of eight avian species provides further insight into their phylogenetic relationships and avian karyotype evolution. Cells 2021, 10, 362.
- Sazanov, A.A.; Romanov, M.N.; Smirnov, A.F. Libraries of large-insert genomic clones as a tool for molecular cytogenetic analysis of avian genome. Russ. J. Genet. 2005, 41, 461–467. https://doi.org/10.1007/s11177-005-0111-z.
- Damas, J.; O’connor, R.; Farré, M.; Lenis, V.P.E.; Martell, H.J.; Mandawala, A.; Fowler, K.; Joseph, S.; Swain, M.T.; Griffin, D.K.; et al. Upgrading short-read animal genome assemblies to chromosome level using comparative genomics and a universal probe set. Genome Res. 2017, 27, 875–884.
- Joseph, S.; O’Connor, R.E.; Al Mutery, A.F.; Watson, M.; Larkin, D.M.; Griffin, D.K. Chromosome level genome assembly and comparative genomics between three falcon species reveals an unusual pattern of genome organisation. Diversity 2018, 10, 113.
- Kretschmer, R.; Furo, I.O.; Gomes, A.J.B.; Kiazim, L.G.; Gunski, R.J.; Garnero, A.D.V.; Pereira, J.C.; Ferguson-Smith, M.A.; de Oliveira, E.H.C.; Griffin, D.K.; et al. A comprehensive cytogenetic analysis of several members of the family Columbidae (Aves, Columbiformes). Genes 2020, 11, 632.
- Kretschmer, R.; Gunski, R.J.; Garnero, A.V.; de Freitas, T.R.O.; Toma, G.A.; Cioffi, M.B.; de Oliveira, E.H.C.; O’Connor, R.E.; Griffin, D.K. Chromosomal analysis in Crotophaga ani (Aves, Cuculiformes) reveals extensive genomic reorganization and an unusual Z-autosome Robertsonian translocation. Cells 2021, 10, 4.
- Furo, I.O.; Kretschmer, R.; O’Brien, P.C.M.; Pereira, J.; Garnero, A.D.V.; Gunski, R.J.; O’Connor, R.E.; Griffin, D.K.; Gomes, A.J.B.; Ferguson-Smith, M.A.; et al. Chromosomal evolution in the phylogenetic context in Neotropical Psittacidae with emphasis on a species with high karyotypic reorganization (Myiopsitta monachus). Front. Genet. 2020, 11, 721.
- Furo, I.O.; Kretschmer, R.; O’Brien, P.C.M.; Pereira, J.; Gunski, R.J.; Garnero, A.D.V.; O’Connor, R.E.; Griffin, D.K.; Ferguson-Smith, M.A.; de Oliveira, E.H.C. Cytotaxonomy of Gallinula melanops (Gruiformes, Rallidae): Karyotype evolution and Phylogenetic inference. Genet. Mol. Biol. 2021, 44.
- Ribas, T.F.A.; Pieczarka, J.C.; Griffin, D.K.; Kiazim, L.G.; Nagamachi, C.Y.; O´Brien, P.C.M.; Ferguson-Smith, M.A.; Yang, F.; Aleixo, A.; O’Connor, R.E. Analysis of multiple chromosomal rearrangements in the genome of Willisornis vidua using BAC-FISH and chromosome painting on a supposed conserved karyotype. BMC Ecol. Evol. 2021, 21, 34.
- Hu, F.; Lin, Y.; Tang, J. MLGO: Phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinform. 2014, 15, 354.
- O’Connor, R.E. Reproductive Isolation, in Individuals and During Evolution, as Result of Gross Genomic Rearrangement in Pigs, Birds and Dinosaurs. Ph.D. Thesis, University of Kent, Canterbury, UK, 2016.
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331.
- de Oliveira, E.H.C.; de Moura, S.P.; dos Anjos, L.J.; Nagamachi, C.Y.; Pieczarka, J.C.; O’Brien, P.C.M.; Ferguson-Smith, M.A. Comparative chromosome painting between chicken and spectacled owl (Pulsatrix perspicillata): Implications for chromosomal evolution in the Strigidae (Aves, Strigiformes). Cytogenet. Genome Res. 2008, 122, 157–162.
- de Oliveira, E.H.C.; Tagliarini, M.M.; dos Santos, M.S.; O’Brien, P.C.M.; Ferguson-Smith, M.A. Chromosome Painting in Three Species of Buteoninae: A Cytogenetic Signature Reinforces the Monophyly of South American Species. PLoS ONE 2013, 8, e70071.
- Nanda, I.; Benisch, P.; Fetting, D.; Haaf, T.; Schmid, M. Synteny conservation of chicken macrochromosomes 1–10 in different avian lineages revealed by cross-species chromosome painting. Cytogenet. Genome Res. 2011, 132, 165–181.
- Furo, I.O.; Monte, A.A.; dos Santos, M.d.S.; Tagliarini, M.M.; O´Brien, P.C.M.; Ferguson-Smith, M.A.; de Oliveira, E.H.C. Cytotaxonomy of Eurypyga helias (Gruiformes, Eurypygidae): First karyotypic description and phylogenetic proximity with Rynochetidae. PLoS ONE 2015, 10, e0143982.
- Kretschmer, R.; Souza, M.S.; Barcellos, S.A.; Degrandi, T.M.; Pereira, J.C.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; Gunski, R.J.; Garnero, A.D.V.; de Oliveira, E.H.C.; et al. Novel insights into chromosome evolution of Charadriiformes: Extensive genomic reshuffling in the wattled jacana (Jacana jacana, Charadriiformes, Jacanidae). Genet. Mol Biol. 2020, 43, e20190236.
- Nanda, I.; Karl, E.; Griffin, D.K.; Schartl, M.; Schmid, M. Chromosome repatterning in three representative parrots (Psittaciformes) inferred from comparative chromosome painting. Cytogenet. Genome Res. 2007, 117, 43–53.
- Furo, I.O.; Kretschmer, R.; O’Brien, P.C.; Ferguson-Smith, M.A.; de Oliveira, E.H.C. Chromosomal diversity and karyotype evolution in South American macaws (Psittaciformes, Psittacidae). PLoS ONE 2015, 10, e0130157.
- Ledesma, M.A.; Cardozo, D.E.; Montalti, D.; Leotta, G.A.; Gunski, R.J. Estudios citogenéticos en tres especies de aves Antárticas. Rev. Cienc. Tecnol. 2005, 7, 68–72.
- Nieto, L.M.; Gunski, R.J. Estudios cromosómicos en atajacaminos (Aves, Caprimulgidae). B. Soc. Biol. Concepción 1998, 69, 161–169.
- de Souza, M.S.; Kretschmer, R.; Barcellos, S.; Costa, A.L.; Cioffi, M.B.; de Oliveira, E.H.C.; Garnero, A.D.V.; Gunski, R.J. Repeat sequence mapping shows different W chromosome evolutionary pathways in two Caprimulgiformes families. Birds 2020, 1, 4.
- Degrandi, T.M.; Garnero, A.D.V.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; Kretschmer, R.; de Oliveira, E.H.C.; Gunski, R.J. Chromosome painting in Trogon s. surrucura (Aves, Trogoniformes) reveals a karyotype derived by chromosomal fissions, fusions, and inversions. Cytogenet. Genome Res. 2017, 151, 208–215.
- de Oliveira, T.D.; Kretschmer, R.; Bertocchi, N.A.; Degrandi, T.M.; de Oliveira, E.H.C.; Cioffi, M.B.; Garnero, A.D.V.; Gunski, R.J. Genomic organization of repetitive DNA in woodpeckers (Aves, Piciformes): Implications for karyotype and ZW sex chromosome differentiation. PLoS ONE 2017, 12, e0169987.


