Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salvatore Saccone | -- | 1852 | 2022-09-26 14:35:08 | | | |
| 2 | Dean Liu | Meta information modification | 1852 | 2022-10-21 03:28:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dugo, K.; Bruno, F.; Sturiale, V.; Brancato, D.; Saccone, S.; Federico, C. Hereditary Transthyretin-Related Amyloidosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/30519 (accessed on 25 July 2026).
Dugo K, Bruno F, Sturiale V, Brancato D, Saccone S, Federico C. Hereditary Transthyretin-Related Amyloidosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/30519. Accessed July 25, 2026.
Dugo, Ketty, Francesca Bruno, Valentina Sturiale, Desiree Brancato, Salvatore Saccone, Concetta Federico. "Hereditary Transthyretin-Related Amyloidosis" Encyclopedia, https://encyclopedia.pub/entry/30519 (accessed July 25, 2026).
Dugo, K., Bruno, F., Sturiale, V., Brancato, D., Saccone, S., & Federico, C. (2022, October 20). Hereditary Transthyretin-Related Amyloidosis. In Encyclopedia. https://encyclopedia.pub/entry/30519
Dugo, Ketty, et al. "Hereditary Transthyretin-Related Amyloidosis." Encyclopedia. Web. 20 October, 2022.
Copy Citation
Point mutations of the transthyretin (TTR) gene are related with hereditary amyloidosis (hATTR), a rare disease whose real incidence is only partially estimated. Somatic mosaicism and other genetic factors influence the expressivity, complexity, progression, and transmission of the disease and should be better investigated, to improve the time to diagnosis and to estimate the real number of cases in endemic and non-endemic areas.
TTR gene
amyloidosis
Gene conversion
1. Introduction
Hereditary amyloidosis TTR-related (hATTR) is a rare disease associated with several point mutations in the transthyretin (TTR) gene, determining abnormal aggregations of the transthyretin protein in different organs; hATTR is often misdiagnosed, as it is characterized by heterogeneous clinical features, including polyneuropathy, cardiomyopathy, vitreous opacity, and nephropathy, common to other diseases [1][2][3][4][5][6][7][8][9]. The history of amyloidosis transthyretin-related (ATTR) was traced back to 1929 to assess the impact of this rare disease in Europe. Reliable data exists, providing a clear picture of the epidemiology in several countries in the European Community. Over time, new diagnostic and therapeutic approaches were adopted, reducing misdiagnosis and contributing to the quality and life expectancy of the affected patients [10][11][12][13].
The most common point mutations of the TTR gene and the closely related clinical features of hATTR have been widely studied. Recently, an abnormal single nucleotide change in the fourth exon was described in a large family in Sicily (Italy) [14]. This was identified by sequence analysis using the Sanger method. Typically, genomic DNA for this type of analysis originates from whole blood, and the genomic DNA can be analyzed by various molecular strategies, not only through direct sequencing, but also using RFLP, southern blot, or other methods, depending on the type and the site of the mutation under analysis. Other tissues, such as buccal cells or hair bulb cells, could be used to obtain genomic DNA, avoiding the more invasive method of blood withdrawal and improving the availability of the patients [14][15][16][17][18][19][20]. The analysis of DNA from tissue other than the blood cells should be performed with caution, as it is possible that somatic mosaicism of the analyzed genomic region can be present in subjects with hATTR [14].
2. Variability and Complexity of hATTR in Sicily (Italy)
The amyloidosis TTR-related symptoms are also determined by unclear mechanisms apart from mutations in the TTR gene and in some other known genes. Since 1:100,000 Italians are affected by this rare disease, further research is necessary to obtain data that are not underpowered [21][22]. In Italy, it is likely that cases of hATTR remain underestimated, as well as some TTR pathological variants being poorly documented in terms of geographical distribution and the clinical characterization of severely affected individuals. Genetic anticipation of serious symptoms, including an early age of onset and the rapid death of the subjects, was described for hereditary amyloidosis related to the Gly47Glu variant of the TTR gene. The genetic analysis showed a transition G>A on exon 2 of the TTR gene, where this point mutation caused the change of glycine with glutamate in position 47. This change seems to alter the normal monomer folding and to promote an easier TTR amyloidotic deposition in the target organs, possibly related with the severity of clinical features. However, other unclear genetic and environmental factors seem to have a role in the onset of severe symptoms of the disease [7][8].
In Italy, hATTR cases have been described in several regions [23]. For example, in Sicily (Italy), three endemic TTR variants were associated with FAP (familial amyloidotic polyneuropathy) (Figure 1 and Figure 2):
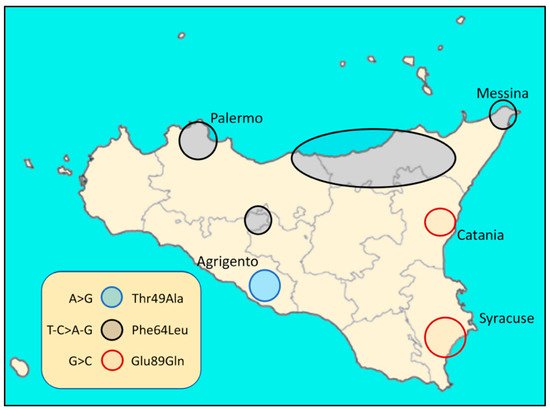
Figure 1. Distribution in Sicily (Italy) of the three endemic TTR variants related to FAP.
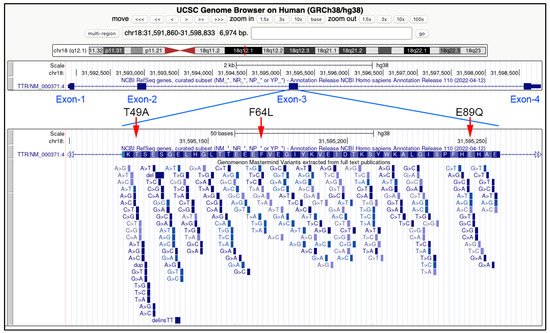
Figure 2. Genomic features of the three endemic TTR variants related to FAP and described in Sicily (Italy). In the upper part of the image, the TTR gene, spanning about 7000 bp in the 18q21.1 chromosomal band, and the position of the four exons. In the bottom panel, an enlargement of the exon three, with the position of the three Sicilian genetic variants: T49A (Thr49Ala), F64L (Phe64), and E89Q (Glu89Gln). All the variants in the third exon described in the scientific literature are indicated below the aminoacidic sequence corresponding to the coding sequence of this exon. Data and images are from the UCSC Genome Browser (http://genome.ucsc.edu, accessed on 26 August 2022).
Glu89Gln (c.325G>C)
Thr49Ala (c.205A>G)
Phe64Leu (c.250 T>C)
To date, all analyzed Sicilian patients were heterozygous for the above mutations and no homozygous TTR variant was found [10][24][25]. However, the presence of rare homozygous individuals for amyloidotic TTR variant cannot be ruled out. Further investigations could clarify this. The preliminary estimate from the first Sicilian hATTR epidemiological study was 8.8/1,000,000 cases, though this value is likely to increase [24][26].
In 1992, a point mutation G>C in exon 3 of the TTR gene was found linked to an aminoacidic change from glutamate (GAG) to glutamine (CAG) in the TTR protein [3]. Previously, a new dominant TTR mutation was found to be related to sensory and peripheral neuropathy, vitreous opacities, and cardiomyopathy, with an early age of onset of disease [27]. In another study, screening tests on serum samples from 74 patients with different TTR mutations and DNA analysis by isoelectric focusing (IEF) showed Glu89Gln and other TTR variants causing hATTR [28]. It was shown that many individuals are heterozygous for Glu89Gln. A founder effect could explain the occurrence of Glu89Gln TTR variants in many Bulgarian individuals, for whom the age of onset is comparable to Sicilian hATTR patients. However, TTR-related cardiomyopathy in Bulgarian patients is worse than that of the Sicilian cohort, and it seems that the clinical phenotype is strongly influenced by respective geographic area. Other TTR pathological variants related to hATTR were described in 2008 in Bulgaria [24][29][30][31][32]. However, further studies could elucidate on the heterogeneity of clinical features and molecular events that occur in modulating the severity and progression of the disease.
3. First Case Report of Somatic Mosaicism in hATTR Patients and Gene Conversion
Recently, a somatic mosaicism was discovered, disclosing a reversion to the normal TTR Glu89Gln (NM_000371.3:c.325G>C) variant. Thus, it would be appropriate to run genomic DNA testing from other tissues in case of negative results from whole blood for all patients with a family health history of hATTR, and it would be interesting to know on what molecular basis somatic mosaicism occurs and how this could affect the clinical features and severity of hATTR disease. Other TTR variants could be identified in genomic DNA from blood, but also from buccal cells or hair of the same patient [14]. The reverting of a pathogenic mutation to normal could represent a new concept of natural gene therapy previously highlighted for other inherited diseases [33]. In humans, it was first demonstrated that a mitotic gene conversion occurred as the genetic mechanism causing revertant mosaicism in a case of a milder clinical phenotype of epidermolysis bullosa [33]. Several mechanisms were proposed to explain a reversion to normal of a DNA sequence, including intragenic recombination, mitotic gene conversion, or second site compensating mutations [34]. A subsequent study supported that double-strand breaks (DSBs) were the initial events of gene conversion and, moreover, how many molecular substrates are necessary for recombinant-repair processes [35]. Understanding gene conversion mechanisms in eukaryotic genome evolution and in inherited diseases can clarify some unclear aspects on the biological answer of an organism to the occurrence of a genetic disease [36][37][38][39]. Gene conversion was demonstrated for the TTR Val30Met variant in vitro and in vivo but is still very far from clinical application [40]. However, it was proposed as a possible gene therapy among the new molecular approaches for treating hATTR [41]. Several known molecular mechanisms can lead to a spontaneous reversion of a gene mutation, causing somatic mosaicism in rare human disorders with more tolerable clinical phenotypes. However, it was highlighted that these phenomena should be sustained by more detailed genetic and molecular analysis, and genomic DNA should be obtained from blood and other tissues, such as buccal mucosa, not forgetting the impact on clinical and ethical aspects [42]. In this regard, several individuals affected by hATTR should be screened to verify the presence of reversion to normality of an amyloidotic TTR gene variant, as observed for Glu89Gln. The clinical impact of this event was not demonstrated but it was observed that a higher rate of individuals from the IV generation did not inherit the Glu89Gln TTR mutation if a parent carried the reversion CAG > GAG in some analyzed tissue sample [14]. Furthermore, in the IV generation, individuals carrying the same form of somatic mosaicism were present in a single family branch.
Somatic mosaicism could influence the transmission of an autosomal trait; however, such could be due to many other issues, including other gene mutations (Figure 3). In 2001, an interesting review showed that somatic mosaicism is very important to evaluate the genetic heterogeneity in several monogenic, neurodegenerative, and complex diseases. More in-depth genetic analyses, including more than one type of tissue, are needed. The human genome is not stable and homogeneous, and this concept is fundamental in the genetic counseling context [14][43][44][45][46].
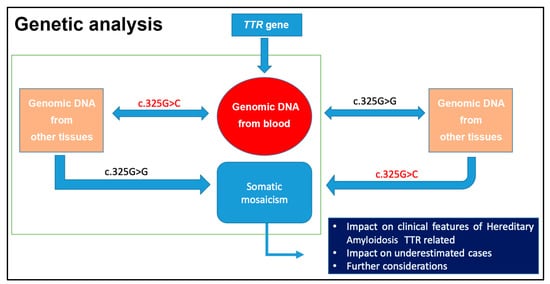
Figure 3. Relevance to analyze genomic DNA from several tissues, as in the case of G>C mutation in the TTR gene (NM_000371.3:c.325G>C) [14].
It has been reported that the expression of the TTR gene varies over time and that there is a difference in the ratios between wild-type and mutated transcripts that often correlate with the age of onset and with the penetrance of the disease. Liver biopsy remains the golden standard for transcriptional analysis of the TTR gene; however, the use of plasma and urine are less invasive methods for relative quantifications between mutant and wild-type transcripts. Wild-type peaks in each sample were used as normalizers and considered to be equal to 1 unit, and the mutated alleles ranged from 0.11 to 1.14 units. In this way, a monoallelic expression on both plasma and urine was surprisingly highlighted. From the processed samples, 87% showed monoallelic expression and 13% biallelic expression in plasma, while in urine 58% showed monoallelic expression and 42% biallelic expression. Furthermore, it was observed that, at an early age, there is a predominant expression of the wild-type form, while the expression of the mutated allele increases with advancing age; when the onset of the disease is around the age of 50, it is possible that there is a biallelic expression, until there is some sort of natural selection or suppression, which favors the expression of the mutated transcripts [47].
The case of a father who passed on p.Glu89Gln to both his twin sons is interesting; one of the two sons presented symptoms before his father and 11 years before his twin. Clearly, additional factors seem to play a fundamental role on the age of onset and the severity of the disease. Investigating the presence of somatic mosaicisms and how these can affect the expression of the TTR gene in different tissues could be useful to understand their role on the expressiveness and on the penetrance of the pathology. Patients with the missense mutation G>C, responsible for the amino acid substitution p.Glu89Gln, in the DNA extracted from blood but not in DNA extracted from buccal or hair bulb cells, have manifested the disease. These patients have not had transcriptional analysis of the TTR gene [14][47][48]. However, the involvement of other unidentified genes in the modulation of TTR gene expression is very likely.
References
- Andrade, C. A peculiar form of peripheral neuropathy: Familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427.
- McCutchen, S.L.; Lai, Z.; Miroy, G.J.; Kelly, J.W.; Colon, W. Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 1995, 34, 13527–13536.
- Almeida, M.D.R.; Ferlini, A.; Forabosco, A.; Gawinowicz, M.; Costa, P.P.; Salvi, F.; Plasmati, R.; Tassinari, C.A.; Altland, K.; Saraiva, M.J. Two transthyretin Variants (TTR Ala-49 and TTR Gln-89) in two Sicilian Kindreds with Hereditary Amyloisosis. Hum. Mutat. 1992, 1, 211–215.
- Lobato, L.; Beirao, I.; Guimaraes, S.M.; Droz, D.; Guimaraes, S.; Grunfeld, J.P.; Noel, L.H. Familial Amyloid Polyneuropathy Type I (Portuguese): Distribution and Characterization of Renal Amyloid Deposits. Am. J. Kidney Dis. 1998, 31, 940–946.
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 2021, 268, 2109–2122.
- Bezerra, F.; Niemietz, C.; Schmidt, H.H.J.; Zibert, A.; Guo, S.; Monia, B.P.; Gonçalves, P.; Saraiva, M.J.; Almeida, M.R. In Vitro and In Vivo Effects of SerpinA1 on the Modulation of Transthyretin Proteolysis. Int. J. Mol. Sci. 2021, 22, 9488.
- Koike, H.; Katsuno, M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines 2019, 7, 11.
- Si, J.-B.; Kim, B.; Kim, J.H. Transthyretin Misfolding, A Fatal Structural Pathogenesis Mechanism. Int. J. Mol. Sci. 2021, 22, 4429.
- Gonzalez-Duarte, A.; Ulloa-Aguirre, A. A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis). Int. J. Mol. Sci. 2021, 22, 13158.
- Parman, Y.; Adams, D.; Obici, L.; Galán, L.; Guergueltcheva, V.; Suhr, O.B.; Coelho, T. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: Where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr. Opin. Neurol. 2016, 29 (Suppl. S1), S3–S13.
- Finsterer, J.; Iglseder, S.; Wanschitz, J.; Topakian, R.; Löscher, W.N.; Grisold, W. Hereditary transthyretin-related amyloidosis. Acta Neurol. Scand. 2019, 139, 92–105.
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2018, 57, 829–837.
- Kim, B.; Ko, Y.H.; Runfola, M.; Rapposelli, S.; Ortore, G.; Chiellini, G.; Kim, J.H. Diphenyl-Methane Based Thyromimetic Inhibitors for Transthyretin Amyloidosis. Int. J. Mol. Sci. 2021, 22, 3488.
- Federico, C.; Dugo, K.; Bruno, F.; Longo, A.M.; Grillo, A.; Saccone, S. Somatic mosaicism wih reversion to normality of a mutated transthyretin allele related to a familial amyloidotic polyneuropathy. Hum. Genet. 2017, 136, 867–873.
- Tsuzuki, T.; Mita, S.; Maeda, S.; Araki, S.; Shimada, K. Structure of the human prealbumin gene. J. Biol. Chem. 1985, 260, 12224–12227.
- Nichols, W.C.; Padilla, L.M.; Benson, M.D. Prenatal Detection of a Gene for Hereditary Amyloidosis. Am. J. Med. Genet. 1989, 34, 520–524.
- Nichols, W.C.; Benson, M.D. Hereditary amyloidosis: Detection of variant prealbumin genes by restriction enzyme analysis of amplified genomic DNA sequences. Clin. Genet. 1990, 37, 44–53.
- Benson, M.D.; Uemichi, T. Transthyretin Amyloidosis. Amyloid. Int. J. Exp. Clin. Investig. 1996, 3, 44–56.
- Carry, B.J.; Young, K.; Fielden, S.; Kelly, M.A.; Sturm, A.C.; Avila, J.D.; Martin, C.L.; Kirchner, H.L.; Fornwalt, B.K.; Haggerty, C.M.; et al. Genomic Screening for Pathogenic Transthyretin Variants Finds Evidence of Underdiagnosed Amyloid Cardiomyopathy From Health Records. Cardio Oncol. 2021, 3, 550–561.
- De Lillo, A.; Pathak, G.A.; De Angelis, F.; Di Girolamo, M.; Luigetti, M.; Sabatelli, M.; Perfetto, F.; Frusconi, S.; Manfellotto, D.; Fuciarelli, M.; et al. Epigenetic profiling of Italian patients identified methylation sites associated with hereditary transthyretin amyloidosis. Clin. Epigenetics 2020, 12, 176.
- Iorio, A.; De Lillo, A.; De Angelis, F.; Di Girolamo, M.; Luigetti, M.; Sabatelli, M.; Pradotto, L.; Mauro, A.; Mazzeo, A.; Stancanelli, C.; et al. Non-coding variants contribute to the clinical heterogeneity of TTR amyloidosis. Eur. J. Hum. Genet. 2017, 25, 1055–1066.
- Kapoor, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Clinical presentation, diagnosis and treatment of TTR amyloidosis. J. Neuromuscul. Dis. 2019, 6, 189–199.
- Pelo, E.; Da Prato, L.; Ciaccheri, M.; Castelli, G.; Gori, F.; Pizzi, A.; Torricelli, F.; Marconi, G. Familial amyloid polyneuropathy with genetic anticipation associated to a gly47glu transthyretin variant in an Italian kindred. Amyloid 2002, 9, 35–41.
- Mazzeo, A.; Russo, M.; Di Bella, G.; Minutoli, F.; Stancanelli, C.; Gentile, L.; Baldari, S.; Carerj, S.; Toscano, A.; Vita, G. Transthyretin-related Familial Amyloid Polyneurophaty (TTRFAP): A single-center Experience in Sicily; an Italian endemic area. J. Neuromuscol. Dis. 2015, 2, S39–S48.
- Kaur, D.; Tiwana, H.; Stino, A.; Sandroni, P. Autonomic neuropathies. Muscle Nerve 2020, 63, 10–21.
- Ferlini, A.; Salvi, F.; Uncini, A.; El-Chami, J.; Winter PAltland, K.; Repetto, M.; Littardi, M.; Campoleoni, A.; Vezzoni, P.; Patrosso, M.C. Homozygosity and heterozygosity for the transthyretin Leu64 mutation: Clinical, biochemical and molecular findings. Clin. Genet. 1996, 49, 10–14.
- Skare, J.C.; Saraiva, M.J.M.; Alves, I.L.; Skare, I.B.; Milunsky, A.; Cohen, A.S.; Skinner, M. A New Mutation Causing Familial Amyloidotic Polyneuropathy. Biochem. Biophys. Res. Commun. 1989, 164, 1240–1246.
- Connors, L.H.; Ericsson, T.; Skare, J.; Jones, L.A.; Lewis, W.D.; Skinner, M. A simple screening test for variant transthyretins associated with familial transthyretin amyloidosis using isoelectric focusing. Biochim. Biophys. Acta 1998, 1407, 185–192.
- Sarafov, S.; Gospodinova, M.; Velina, V.G.; Kirov, A.; Teodora, C.; Todorov, T.; Todorova, A.; Tournev, I. Epidemiology of Familial Amyloid Polyneuropathy in Bulgaria. Orphanet J. Rare Dis. 2015, 10 (Suppl. S1), O2.
- Gagliardi, C.; Gospodinova, M.; Longhi, S.; Milandri, A.; Cinelli, M.; Tournev, I.; Salvi, F.; Rapezzi, C. Glu89Gln transthyretin-related amyloidosis in Italy and Bulgaria: Does geographic area influence phenotype beyond the shared mutation? Orphanet J. Rare Dis. 2015, 10 (Suppl. S1), P23.
- Kirov, A.; Sarafov, S.; Pavlova, Z.; Todorov, T.; Chamova, T.; Gospodinova, M.; Tournev, I.; Mitev, V.; Albena Todorova, A. Founder effect of the Glu89Gln TTR mutation in the Bulgarian population. Amyloid 2019, 26, 181–185.
- Pavlova, Z.; Sarafov, S.; Todorov, T.; Kirov, A.; Chamova, T.; Gospodinova, M.; Tournev, I.; Mitev, V.; Todorova, A. Characterization of population genetic structure of hereditary transthyretin amyloidosis in Bulgaria. Amyloid 2021, 28, 219–225.
- Jonkman, M.F.; Scheffer, H.; Stulp, R.; Pas, H.H.; Nijenhuis, M.; Heeres, K.; Owaribe, K.; Pulkkinen, L.; Uitto, L. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene Conversion. Cell 1997, 88, 543–551.
- Hirshhorn, R. In vivo reversion to normal of inherited mutations in humans. J. Med. Genet. 2003, 40, 721–728.
- Chuzhanova, N.; Chen, J.M.; Bacolla, A.; Patrinos, G.P.; Férec, C.; Wells, R.D.; Cooper, D.N. Gene Conversion Causing Human Inherited Disease: Evidence for Involvement of Non-B-DNA-Forming Sequences and Recombination-Promoting Motifs in DNA Breakage and Repair. Hum. Mutat. 2009, 30, 1189–1198.
- Paques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404.
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271.
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double strand break repair model for recombination. Cell 1983, 33, 25–35.
- Chen, J.M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762–775.
- Nakamura, M.; Ando, Y.; Nagahara, S.; Sano, A.; Ochiya, T.; Maeda, S.; Kawaji, T.; Ogawa, M.; Hirata, A.; Terazaki, H. Targeted conversion of the transthyretin gene in vitro and in vivo. Gene Ther. 2004, 11, 838–846.
- Ueda, M.; Ando, Y. Recent advances in transthyretin amyloidosis therapy. Transl. Neurodegener. 2014, 3, 19.
- Youssoufian, H.; Pyeritz, R.E. Mechanisms and Consequences of Somatic Mosaicism in Humans. Nat. Rev. Genet. 2002, 3, 748–758.
- Gottlieb, B.; Beitel, L.K.; Trifiro, M.A. Somatic Mosaicism and Variable Expressivity. Trend Genet. 2001, 17, 79–82.
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392.
- Lopes, L.R.; Futema, M.; Mohammed, M.; Akhtar, M.M.; Lorenzini, M.; Pittman, A.; Syrris, P.; Elliott, P.M. Prevalence of TTR variants detected by whole-exome sequencing in hypertrophic cardiomyopathy. Amyloid 2019, 26, 243–247.
- Patel, J.K.; Rosen, A.M.; Chamberlin, A.; Feldmann, B.; Antolik, C.; Zimmermann, H.; Johnston, T.; Narayana, A. Three Newly Recognized Likely Pathogenic Gene Variants Associated with Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2022, 1–13.
- Yordanova, I.; Pavlova, Z.; Kirov, A.; Todorov, T.; Alexiev, A.; Sarafov, S.; Mateva, L.; Chamova, T.; Gospodinova, M.; Mitev, V. Monoallelic expression of the TTR gene as a contributor to the age at onset and penetrance of TTR-related amyloidosis. Gene 2019, 705, 16–21.
- Niemietz, C.; Bezerra, F.; Almeida, M.R.; Guo, S.; Monia, B.P.; Saraiva, M.J.; Schütz, P.; Schmidta, H.H.-J.; Zibert, A. SERPINA1 modulates expression of amyloidogenic transthyretin. Exp. Cell Res. 2020, 395, 112217.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
21 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No