 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dean Liu | -- | 2186 | 2022-10-19 01:38:04 |
Video Upload Options
Ensembl Genomes is a scientific project to provide genome-scale data from non-vertebrate species. The project is run by the European Bioinformatics Institute, and was launched in 2009 using the Ensembl technology. The main objective of the Ensembl Genomes database is to complement the main Ensembl database by introducing five additional web pages to include genome data for bacteria, fungi, invertebrate metazoa, plants, and protists. For each of the domains, the Ensembl tools are available for manipulation, analysis and visualization of genome data. Most Ensembl Genomes data is stored in MySQL relational databases and can be accessed by the Ensembl Perl API, virtual machines or online. Ensembl Genomes is an open project, and most of the code, tools, and data are available to the public. Ensembl and Ensembl Genomes software uses a "permissive Apache-style open-source" license, making it free for all users.
1. Displaying Genomic Data
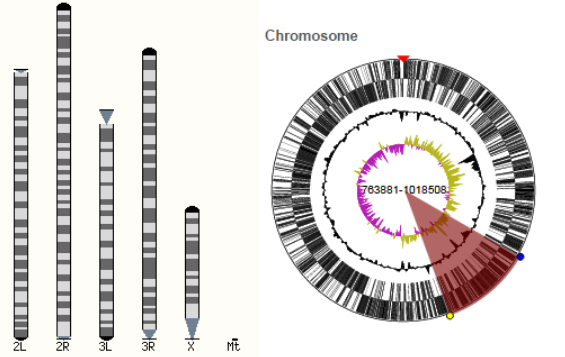
The key feature of Ensembl Genomes is its graphical interface, which allows users to scroll through a genome and observe the relative location of features such as conceptual annotation (e.g. genes, SNP loci), sequence patterns (e.g. repeats) and experimental data (e.g. sequences and external sequence features mapped onto the genome).[1] Graphical views are available for varying levels of resolution from an entire karyotype, down to the sequence of a single exon. Information for a genome is spread over four tabs, a species page, a ‘Location’ tab, a ‘Gene’ tab and a ‘Transcript’ tab, each providing information at a higher resolution.
Searching for a particular species using Ensembl Genomes redirects to the species page. Often, a brief description of the species is provided, as well as links to further information and statistics about the genome, the graphical interface and some of the tools available.
A karyotype is available for some species in Ensembl Genomes.[2] If the karyotype is available there will be a link to it in the Gene Assembly section of the species page. Alternatively if users are in the ‘Location’ tab they can also view the karyotype by selecting ‘Whole genome’ in the left-hand menu. Users can click on a location within the karyotype to zoom in to one specific chromosome or a genomic region.[2] This will open the ‘Location’ Tab.
In the 'Location' tab, users can browse genes, variations, sequence conservation, and other types of annotation along the genome.[3] The 'Region in detail' is highly configurable and scalable, and users can choose what they want to see by clicking on the 'Configure this page' button at the bottom of the left-hand menu. By adding and removing tracks users will be able to select the type of data they want to have included in the displays.[3] Data from the following categories can be easily added or removed from this 'Location' tab view: 'Sequence and assembly', 'Genes and transcripts', 'mRNA and protein alignments', 'Other DNA alignments', 'Germline variation', 'Comparative genomics', among others.[3] Users can also change the display options such as the width.[3] A further option allows users to reset the configuration back to the default settings.[3]
More specific information about a select gene can be found in the ‘Gene’ tab. Users can get to this page by searching for desired gene in the search bar and clicking on the gene ID or by clicking on one of the genes shown in the ‘Location’ tab view. The ‘Gene’ tab contains gene-specific information such as gene structure, number of transcripts, position on the chromosome and homology information in the form of gene trees.[4] This information can be accessed via the menu on the left-hand side.
A 'Transcript' tab will also appear when a user chooses to view a gene. The 'Transcript' tab contains much of the same information as the 'Gene' tab, however it is focused on only one transcript.[4]
2. Tools
2.1. Adding Custom tracks to Ensembl Genomes
Ensembl Genomes allows comparing and visualising user data while browsing karyotypes and genes. Most Ensembl Genomes views include an ‘Add your data’ or ‘Manage your data’ button that will allow the user to upload new tracks containing reads or sequences to Ensembl Genomes or to modify data that has been previously uploaded.[5] The uploaded data can be visualised in region views or over the whole karyotype. The uploaded data can be localised using Chromosome Coordinates or BAC Clone Coordinates.[6] The following methods can be used to upload a data file to any Ensembl Genomes page:[7]
- Files smaller than 5 MB can be either uploaded directly from any computer or from a web location (URL) to the Ensembl servers.
- Lager files can only be uploaded from web locations (URL).
- BAM files can only be uploaded using the URL-based approach. The index file (.bam.bai) should be located in the same webserver.
- A Distributed Annotation System source can be attached from web locations.
The following file types are supported by Ensembl Genomes:[8]

- BED
- BedGraph
- Generic
- GFF/GTF
- PSL
- WIG
- BAM
- BigBed
- BigWig
- VCF
The data is uploaded temporarily into the servers. Registered users can log in and save their data for future reference. It is possible to share and access the uploaded data using and an assigned URL.[9] Users are also allowed to delete their custom tracks from Ensembl Genomes.
2.2. BioMart
BioMart is a programming free search engine incorporated in Ensembl and Ensembl Genomes (except for Ensembl Bacteria) for the purpose of mining and extracting genomic data from the Ensembl databases in table formats like HTML, TSV, CSV or XLS.[10] Release 45 (2019) of Ensembl Genomes has the following data available at the BioMarts:
- Ensembl Protists BioMart: includes 33 species and variations for Phytophthora infestans and Phaeodactylum tricornutum[11]
- Ensembl Fungi BioMart: includes 56 species and variations for Fusarium graminearum, Fusarium oxysporum, Schizosaccharomyces pombe, Puccinia graminis, Verticillium dahliae, Zymoseptoria tritici, and Saccharomyces cerevisiae[12]
- Ensembl Metazoa BioMart: includes 78 species and variations for Aedes aegypti, Anopheles gambiae and Ixodes scapularis[13]
- Ensembl Plants: includes 67 species and variations for Arabidopsis thaliana, Brachypodium distachyon, Hordeum vulgare, Oryza glaberrima, Oryza glumipatula, Oryoza sativa indica, Oryza sativa japonica, Solanum lycopersicum, Sorghum bicolor, Triticum aestivum, Vitis vinifera, and Zea mays[14]
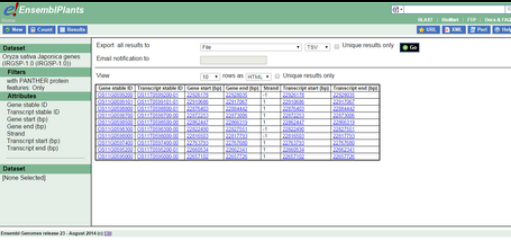
The purpose of the BioMarts in Ensembl Genomes is to allow the user to mine and download tables containing all the genes for a single species, genes in a specific region of a chromosome or genes on one region of a chromosome associated with an InterPro domain.[15] The BioMarts also include filters to refine the data to be extracted and the attributes (Variant ID, Chromosome name, Ensembl ID, location, etc.) that will appear in the final table file can be selected by the user.
The BioMarts can be accessed online in each corresponding domain of Ensembl Genomes or the source code can be installed in UNIX environment from the BioMart git repository[16]
2.3. BLAST
A BLAST interface is provided to allow users to search for DNA or protein sequences against the Ensembl Genomes. It can be accessed by the header, located on top of all Ensembl Genome pages, titled BLAST. The BLAST search can be configured to search against individual species or collections of species (maximum of 25). There is a taxonomic browser to allow the selection of taxonomically related species.[17]
2.4. Sequence Search
Ensembl Genomes provides a second sequence search tool, that uses an algorithm based on Exonerate, that is provided by European Nucleotide Archive.[17] This tool can be accessed by the header, located on top of all Ensembl Genome pages, titled Sequence Search. Users can then choose whether they would like Exonerate to search against all species in the Ensembl Genomes division or against all species in Ensembl Genomes. They can also choose the 'Maximum E-value', which will limit the results that appear to those with E-values below the maximum. Finally users can choose to use an alternative search mode by selecting 'Use spliced query'.
2.5. Variant Effect Predictor
The Variant Effect Predictor is one of the most used tools in Ensembl and Ensembl Genomes. It allows to explore and analyse what is the effect that the variants (SNPs, CNVs, indels or structural variations) have on a particular gene, sequence, protein, transcript or transcription factor.[18] To use VEP, the users must input the location of their variants and the nucleotide variations to generate the following results:[19]
- Genes and transcripts affected by the variant
- Location of the variants
- How the variant affects the protein synthesis (e.g. generating a stop codon)
- Comparison with other databases to find equal known variants
There are two ways in which the users can access the VEP. The first form is online-based. In this page, the user generates an input by selection the following parameters:[20]
- Species to be compared. The default database for comparison is Ensembl Transcripts, but for some species, other sources can be selected.
- Name for the uploaded data (this is optional, but it will make easier to identify the data if many VEP jobs have been performed)
- Selection of the input format for the data. If an incorrect file format is selected, VEP will throw an error when running.
- Fields for data upload. Users can upload data from their computers, from an URL-based location or by copying directly their contents into a text box.
Data upload to VEP supports VCF, pileup, HGVS notations and a default format.[21] The default format is a whitespace-separated file that contains the data in columns. The first five columns indicate the chromosome, start location, end location, allele (pair of alleles separated by a '/', with the reference allele first) and the strand (+ for forward or – for reverse).[22] The sixth column is a variation identifier and it is optional. If it is left in blank, VEP will assign an identifier to in output file.
VEP also provides additional identifier options to the users, extra options to complement the output and filtering.[23] The filtering options allow features like removal of known variants from results, returning variants in exons only, and restriction of results to specific consequences of the variants.[24]
VEP users also have the possibility of viewing and manipulating all the jobs associated with their session by browsing the "Recent Tickets" tab. I this tab the users can view the status of their search (success, queued, running or failed) and save, delete or resubmit jobs.[25]
The second option to use VEP is by downloading the source code for its use in UNIX environments.[26] All the features are equal between the online and script versions. VEP can also be used with online instances like Galaxy.
When a VEP job is completed the output is a tabular file that contains the following columns:[27]
- Uploaded variation - as chromosome_start_alleles
- Location - in standard coordinate format (chr:start or chr:start-end)
- Allele - the variant allele used to calculate the consequence
- Gene - Ensembl stable ID of affected gene
- Feature - Ensembl stable ID of feature
- Feature type - type of feature. Currently one of Transcript, RegulatoryFeature, MotifFeature.
- Consequence - consequence type of this variation
- Position in cDNA - relative position of base pair in cDNA sequence
- Position in CDS - relative position of base pair in coding sequence
- Position in protein - relative position of amino acid in protein
- Amino acid change - only given if the variation affects the protein-coding sequence
- Codon change - the alternative codons with the variant base in upper case
- Co-located variation - known identifier of existing variation
- Extra - this column contains extra information as key=value pairs separated by ";". Displays extra identifiers.

Other common output formats for VEP include JSON and VDF formats.[28]
2.6. Data Access and Virtual Machines
The data contained in Ensembl Genomes can be accessed using a Core API based in PEARL that generates interfaces for accessing Ensembl Genomes data.
Additionally, virtual machines based in VirtualBox are made available by Ensembl Genomes. This virtual machines are pre-configured and can be set to browse any domain of Ensembl Genomes.[29]
Web instances like the ones used in Galaxy or Amazon Elastic Compute Cloud can also be used to access Ensembl features.
3. Current Species
Ensembl Genomes makes no attempt to include all possible genomes, rather the genomes that are included on the site are those that are deemed to be scientifically important. Each site contains the following number of species:
- The bacterial division of Ensembl now contains all bacterial genomes that have been completely sequenced, annotated and submitted to the International Nucleotide Sequence Database Collaboration (European Nucleotide Archive, GenBank and the DNA Database of Japan). The current dataset contains 44,048 genomes.[30]
- Ensembl Fungi contains 1014 genomes[31]
- Ensembl Metazoa contains 78 genomes for invertebrate species.[32] The main Ensembl site contains 236 genomes for vertebrate species.[33]
- Ensembl Plants contains 67 genomes[34]
- Ensembl Protists contains 237 genomes[35]
4. Collaborations
Ensembl Genomes continuously expands the annotation data through collaboration with other organisations involved in genome annotation projects and research. The following organisations are collaborators of Ensembl Genomes:[36]
- AllBio
- Barley
- Culicoides sonorensis
- Gramene
- INFRAVEC
- Microme
- PomBase
- PhytoPath
- transPLANT
- Triticeae Genomics for Sustainable Agriculture
- VectorBase
- Wheat Rust Genomic Improvement
- WormBase
- WormBase ParaSite
References
- Kersey, P. J.; Staines, D. M.; Lawson, D.; Kulesha, E.; Derwent, P.; Humphrey, J. C.; Hughes, D. S. T.; Keenan, S. et al. (2011). "Ensembl Genomes: An integrative resource for genome-scale data from non-vertebrate species". Nucleic Acids Research 40 (Database issue): D91–D97. doi:10.1093/nar/gkr895. PMID 22067447. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3245118
- "Whole Genome". Ensembl Genomes. http://fungi.ensembl.org/Help/View?id=154. Retrieved 7 September 2014.
- "Frequently Asked Questions". Ensembl Genomes. http://ensemblgenomes.org/node/30399. Retrieved 7 September 2014.
- Spudich, G; Fernández-Suárez, X. M.; Birney, E (2007). "Genome browsing with Ensembl: A practical overview". Briefings in Functional Genomics and Proteomics 6 (3): 202–19. doi:10.1093/bfgp/elm025. PMID 17967807. https://dx.doi.org/10.1093%2Fbfgp%2Felm025
- "Uploading your data to Ensembl". Ensembl Genomes. http://ensemblgenomes.org/info/access/using-your-own-data. Retrieved 9 September 2014.
- "Coordinates for data location in Ensembl Genomes". Ensembl Genomes. http://plants.ensembl.org/info/website/upload/coords.html#bacclone. Retrieved 9 September 2014.
- "Methods for data upload". Ensembl Genomes. http://plants.ensembl.org/info/website/upload/index.html#formats. Retrieved 9 September 2014.
- "Supported data files". Ensembl Genomes. http://plants.ensembl.org/info/website/upload/index.html#formats. Retrieved 9 September 2014.
- "Saving and Sharing data in Ensembl Genomes". Ensembl Genomes. http://plants.ensembl.org/info/website/upload/index.html#formats.
- "Data Mining in Ensembl with Data Mining in Ensembl with BioMart". 2014. p. 2. http://asia.ensembl.org/info/website/tutorials/mining_with_biomart.pdf. Retrieved 11 September 2014.
- "Ensembl Protists". Ensembl Genomes. http://protists.ensembl.org/biomart/martview/b5a124953d0d5abc16130408a00a0195. Retrieved 1 October 2019.
- "Ensembl Fungi". Ensembl Genomes. http://fungi.ensembl.org/biomart/martview/5b1b1f7a1f6a2367f491c6b553f6aa60. Retrieved 1 October 2019.
- "Ensembl Metazoa". Ensembl Genomes. http://metazoa.ensembl.org/biomart/martview/ef188d8b8765e906ca1f6267a836a3c1. Retrieved 1 October 2019.
- "Ensembl Plants". Ensembl Genomes. http://plants.ensembl.org/biomart/martview/300ab140234f173a3bcd1f77fa14f251. Retrieved 1 October 2019.
- "Data Mining in Ensembl with Data Mining in Ensembl with BioMart". 2014. p. 3. http://asia.ensembl.org/info/website/tutorials/mining_with_biomart.pdf. Retrieved 11 September 2014.
- "BioMart 0.9.0 user manual". May 2014. p. 5. http://www.biomart.org/other/biomart_0.9_0_documentation.pdf. Retrieved 11 September 2014.
- "Frequently Asked Questions". Ensembl Genomes. http://ensemblgenomes.org/node/32859l. Retrieved 11 September 2014.
- "Variant Effect Predictor". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/index.html. Retrieved 11 September 2014.
- "Variant Effect Predictor results overview". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/index.html. Retrieved 11 September 2014.
- "Data input to VEP". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/online/input.html#ident. Retrieved 11 September 2014.
- "VEP supported file formats". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/vep_formats.html. Retrieved 11 September 2014.
- "VEP default file". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/vep_formats.html. Retrieved 11 September 2014.
- "VEP options and extras". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/online/input.html#ident. Retrieved 11 September 2014.
- "VEP filtering". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/online/input.html#ident. Retrieved 11 September 2014.
- "VEP jobs". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/online/input.html#ident. Retrieved 11 September 2014.
- "VEP script download". Ensembl. http://asia.ensembl.org/info/docs/tools/vep/script/vep_download.html#windows. Retrieved 11 September 2014.
- "VEP Output". Ensembl Genomes. http://asia.ensembl.org/info/docs/tools/vep/script/vep_download.html#windows. Retrieved 11 September 2014.
- "VEP Output formats". Ensembl Genomes. http://asia.ensembl.org/info/docs/tools/vep/script/vep_download.html#windows. Retrieved 11 September 2014.
- "Virtual Machines". Ensembl Genomes. http://ensemblgenomes.org/info/access/virtual_machine. Retrieved 11 September 2014.
- "Species List". Ensembl Genomes. http://bacteria.ensembl.org/species.html. Retrieved 1 October 2019.
- "Species List". Ensembl Genomes. http://fungi.ensembl.org/species.html. Retrieved 1 October 2019.
- "Species List". Ensembl Genomes. http://metazoa.ensembl.org/species.html. Retrieved 1 October 2019.
- "Species List". Ensembl Genomes. http://asia.ensembl.org/info/about/species.html. Retrieved 1 October 2019.
- "Species List". Ensembl Genomes. https://plants.ensembl.org/species.html. Retrieved 1 October 2019.
- "Species List". Ensembl Genomes. http://protists.ensembl.org/species.html. Retrieved 1 October 2019.
- "Collaborators - Ensembl Genomes". Ensembl Genomes. http://ensemblgenomes.org/info/about/collaborations. Retrieved 3 September 2014.

