 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elvira Sgobba | + 2223 word(s) | 2223 | 2020-11-06 07:16:51 | | | |
| 2 | Catherine Yang | -9 word(s) | 2214 | 2020-11-16 06:53:29 | | |
Video Upload Options
MALDI Mass Spectrometry is an analytical technique able to study the complex interdependency between chemical interaction and structural conformation of several protein based assemblies.
1. Introduction
Matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) was the first time developed by Hillekanmp and Karas [1]. Briefly, the irradiation of a laser with a specific wavelength on a solid sample, covered with a small UV absorbing molecule, leads to the production of gaseous ions, that are analyzed based on their mass-to-charge ratio (m/z) by a mass spectrometer [2]. This technique is applied to several scientific fields from chemistry to biomedicine because can detect a broad variety of compounds such as small molecules until large proteins using a very small amount of sample. MALDI-MS is commonly used in proteomics to identify proteins and their post-translational modifications by detection of tryptic peptides and its comparison to specific databases [3][4]. On the other hand, the soft nature of the MALDI allows also us to analyze entire protein complexes that are bound by noncovalent interactions [5]. Understanding noncovalent interactions between proteins are of great interest and crucial for the biological function such as enzymatic catalysis [6], modulation of signal transduction [7], and immunomodulation [8]. In this regard, the conformation of the protein has a strong effect on the noncovalent interactions and therefore on biological effects [9]. The information regarding the influence of the conformation in the interaction and vice versa is a great interest not only for basic research or structural biology but also in the pharmaceutical fields in which protein-based drugs are continuously developed e.g., vaccines [10]. The interaction of antigen and antibody is a great example of how the conformation of the antigen is crucial for its interaction [11][12]. Therefore, noncovalent interactions are involved in the functionality of a complex protein network [13]. To analyze this complex and sensitive information, sophisticated methods are applied to preserve the conformational and reactivity of the protein, as shown in Figure 1.
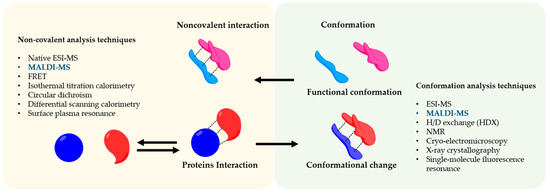
Figure 1. Noncovalent Protein-protein assembly. The picture shows noncovalent interactions in protein-protein complexes and an overview of the techniques to study their conformation and interaction.
2. MALDI Mass Spectrometry Analysis as an Approach to Unravel Interactions in Protein Assembly Guided by Molecular Conformation
Mass spectrometry is a technique able to measure the m/z of several ionized compounds that are analyzed through the use of a mass analyzer and highly sensitive detectors giving several advantages in analytical procedures such as high specificity, sensitivity, speed, and stoichiometry [14][15]. In general, a mass spectrometer consists of three components as schematically shown in Figure 2.
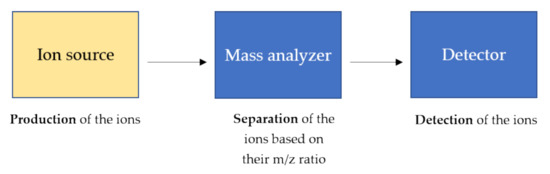
Figure 2. General components of a mass spectrometer. The ions, produced by the ion source, are separated based on their m/z ratio and detected by high sensitive detectors. The ion source produces the ions in the gas phase from the sample. In the mass analyzer, such as triple quadrupole and time-of-flight (TOF), the ions are separated based on the m/z charge ratio and at the end, the ionic currents are detected by conversion to electrical signals in the detector.
The ion source is the main component and the heart of the mass spectrometer in which the analytes are ionized through specific and diverse ionization processes, and therefore detectable by the mass spectrometer. The parameters of the ion source and the sample preparation strongly affect the energy stability of noncovalent interactions, that can be preserved during the ionization process in the gas phase [16]. Therefore, the ion source and its ionization process strongly affect the sensitivity of the noncovalent structures such as the tertiary and quaternary structures of proteins.
Soft ionization processes, such as electrospray ionization (ESI) and MALDI, are employed for structural analysis of the protein elucidating not only noncovalent interactions but also conformational changes of the proteins. For such purpose, several techniques such as hydrogen/deuterium exchange (HDX), native MS, limited proteolysis, cross-linking, and fast photochemical derivatizations are applied to investigate the relationship function and conformation [17][18].
Although Electrospray Ionization (ESI) is the preferred method to study conformations and noncovalent interactions between proteins [19], MALDI-MS remains a simple and fast alternative to study protein-protein complexes. Even though the high energy of the laser, MALDI can preserve noncovalent interactions [5], and yields stoichiometric information of the protein-protein complex [17]. A general advantage of implementing MALDI in analytical procedures is also a user-friendly sample preparation (small sample volumes, no complicated protocols, and high salt tolerance), compatible with high-throughput screening procedures [20] and clinical applications [21]. On the other hand, special and expensive detectors such as ion conversion dynode (ICD), which is the only commercially available high-mass detector, are required for the detection of intact protein-protein complexes with very good sensitivity [22]. Another disadvantage of MALDI-MS to take into account is the challenging in the performance of absolute quantification due to sample composition, sample morphology, inhomogeneous matrix crystallization, laser conditions and, depletions during laser exposure [23]. This review is going to describe MALDI-MS-based procedures for finding functional conformations of a protein involved in a protein-protein complex as described in Figure 3.
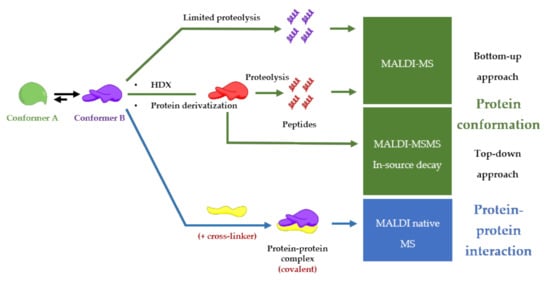
Figure 3. Conformation and protein-protein interaction study based on MALDI-mass spectrometry finding the functional conformation.
2.1. Limited Proteolysis MALDI-MS
Limited proteolysis mass spectrometry is a trend method to study conformational effects upon interaction with small molecules [24]. The limited proteolysis is restricted digestion of the protein with a low concentration of proteases. It leads to controlled proteolysis at putative cleavage sites, based on the backbone plasticity and/or local unfolding of the protein region [25].
A combination of limited proteolysis and MALDI-MS was applied for the first time to study a multi-component biomolecular assembly DNA-protein [26] and to perform structural studies using proteolytic footprinting [27]. Similar approaches were applied to protein-protein interactions between cell cycle regulatory proteins, p21 and Cdk2 [28][29], or on capsid viral protein complexes to elucidate mobile features of the viral proteins [30] e.g., autocatalytically cleavable protein α in Flock House virus [31]. The activation-dependent conformational changes of ß-arrestins are revealed using limited proteolysis in combination with both sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry analysis. This mass spectrometry-based method can be adapted as a tool to study the nature of ß-arrestins conformational changes downstream of different receptors, as well as to study how each conformational change can be associate with a specific ß-arrestin function e.g., desensitization, internalization, and signaling [32][33]. The structure-function dependent interaction between Hsp90 and its co-chaperone Cdc37 resulting in the facilitation of folding and activation of numerous proteins. Such interaction was studied by limited proteolysis in conjunction with MALDI-TOF-MS analysis of peptide fragments and peptide microsequencing [34]. Another example is given by epitope analysis of antibody-antigen complexes performed also by limited proteolysis coupled with MALDI-MS [35] as shown the Figure 4.
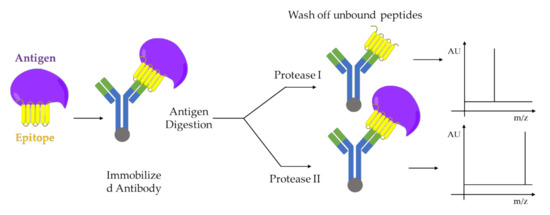
Figure 4. Epitope analysis by limited proteolysis-MALDI-MS approach. Two proteases I and II, which cleaves the antigen at different sites, are used for digestion. The immobilized antibody-epitope complex can be analyzed directly by MALDI-MS. Adapted from [35].
2.2. HDX-MALDI-MS
HDX is a powerful labeling tool that investigates protein conformational alteration by monitoring differential deuterium incorporation in altered regions [36]. Protein hydrogens attached to electronegative atoms (e.g., amide hydrogens) continuously exchange with solvent hydrogen and deuterium either in an acid or base-catalyzed manner [37][38]. The measurement of the exchange rate for these amides provides detailed information about their surrounding environments [39][40]. In HDX-MS there are two ways to measure the exchange rate of the amides: 1) by measure the m/z ratio of the peptides produced after proteolysis of deuterated protein (“bottom-up approach”) and 2) by measure the m/z ratio of fragments produced by ion dissociation, for example, electron-capture dissociation (ECD) or electron-transfer dissociation (ETD), of the deuterated protein avoiding randomization of deuterons within proteins in the gas phase (“top-down approach”) [36]. An interesting application of HDX coupled with mass spectrometry and NMR spectroscopy consists of studying the conformational and dynamic changes of an allosteric system, consisting of a transition of the system within several conformational states [41]. HDX-MShas enabled the investigation of protein folding intermediates, the characterization of transient structure in intrinsically disordered protein [42].
An example of the bottom-up approach was reported by Mandel et al., where the combination of HDX-MALDI-MS was employed to identify peptic fragments from protein complexes at the interacting interface between kinase inhibitors and cyclic-AMP-dependent protein kinase. This complex retains deuterium under hydrogen exchange conditions due to decreased solvent accessibility at the interface of the complex [43][44]. The isotope exchange of polypeptide backbone amide hydrogens of hemoglobin (Hb) and following tryptic digestion was carried out inside red blood cells and monitored using MALDI-MS to explore the conformational transition associated with oxygenation [45]. Conformational dynamics of factor XIII activation induced by calcium were also investigated by HDX-MALDI-MS of tryptic peptides [46]. In this context, limited proteolysis combined with HDX and quantitative LC-MALDI-MS was employed to monitor the conformational changes of troponin C after binding with ligand calcium [47].
On the other hand, a HDX-MALDI-MS using a top-down approach trough in-source decay analysis of deuterated cytochrome c yielded an extensive series of c-fragment ions that originate from the cleavage of nearly all N-Cα bonds allowing for a detailed analysis of the deuterium content of the backbone amides with a minimal hydrogen scrambling. In this way, the dynamic behavior of cytochrome c in solution is accurately reflected in the deuterium content of the fragment ions [48].
Moreover differential and native HDX-MALDI-MS semiautomated conformational screening was performed by changing the polarity of the solvent and different conformations of the protein were evaluated [49]. In detail, the small size proteins, ubiquitin, and insulin were analyzed by using a nonaqueous matrix solution (5 mg/mL of sinapic acid in acetonitrile with 0.1% 3-nitrobenzyl alcohol) to minimize the D/H back exchange. Spatially resolved deuteration levels are readily obtained by mass analysis of consecutive fragment ions. This semi-automated approach is an important step forward in the automation of proteomics approaches that are ongoing developing for clinical and biomedical applications [50].
The coupling of MALDI-MS to HDX is limited by undesired back-exchange reactions from the air humidity, but optimized protocols were developed to minimize this issue in sample preparation [51]. For example, the low temperature of the sample plate minimizes the amount of back exchange [45].
2.3. Protein Derivatization
Since chemical modification has a conformational impact on protein, protein derivatization is another approach to study the functional conformation of a protein [52][53]. The yield of these derivatizations depends on the spatial localization of amino acids in the protein and therefore on the protein folding. In MALDI-MS, the bottom-up approach of derivatized protein is employed for conformational studies and native MS. The top-down approach for the conformational study is still carried out with ESI coupled mass spectrometers in which infrared multiple photon dissociation (IRMPD) and electron capture/electron transfer dissociation (ECD/ETD) are employed to dissociate the derivatized protein and to analyze the cross-linked fragments. On the other hand, through in-source decay, a top-down approach can be carried out by MALDI-MS, but so far, an established method is not reported with derivatized protein. Moreover, the protein derivatization is employed to convert noncovalent protein-protein complex, such as integral membrane protein complexes, to covalent, and therefore the resulting covalent complex is detected by MALDI-MS [54]. One of the chemical modifications mostly used in MALDI-MS is cross-linking (CL). The concept of protein cross-linking in combination with mass spectrometry holds great promise to derive structural information on protein conformation, protein-protein interactions, and protein interaction networks in complex biological samples [55][56]. The general workflow based on MALDI is the detection of peptide derivates from cross-linked proteins with a specific conformation and/or specific interaction with another protein using MALDI-MS/MS and homo-bifunctional amino group-specific [57] and isotope-labeled cross-linkers [58].
Hundreds of chemicals described in the literature or commercially available are based on a limited number of reactions [59][60]. Examples of cross-linkers used in MALDI-MS are amino-reactive cross-linkers (N-hydroxysuccinimide esters) [61], sulfhydryl-reactive cross-linkers, maleic acid imides, disuccinimidyl suberate, bis(sulfosuccinimidyl suberate and 1,1′-(suberoyldioxy) bis azabenzotriazole [62], glutaraldehyde [52][63] and photo-reactive cross-linkers [64][65][66].
Promising for future applications in structural proteomics and imaging is the development of a derivatization method based on nanosecond photoreactions of neuropeptides using nitrobenzaldehyde giving the reactive 2-nitrosobenzoic anion able to react with the primary group of a protein [67]. By combining the use of bisthiopropionic acid N-succinimidyl ester (BipS), a photocleavable, isotopically coded, fluorescent cross-linker, the first direct evidence was provided for the docking site of a phosphorylated G-protein-coupled receptor C terminus on the multifunctional adaptor protein beta-arrestin, clearly demonstrating the broad potential and application in structural and cellular biology [68]
2.4. MALDI Native Mass Spectrometry
First studies reported that the observation of entire noncovalent complexes, without using any derivatization procedures, in high mass MALDI-MS is possible only by acquiring the mass spectra by the first few laser shots at a new sample spot (“first shot phenomenon”) [69][70][71].
Recently, Beaufour et al. developed a method for native mass spectrometry of proteinsbased on liquid deposits. The molecular arrangement between matrix and complex is involved in the stability of noncovalent interaction during the MALDI events. In contrast to the solid deposit that has an irreversible frozen arrangement, in liquid deposit, regeneration of an ideal rearrangement following a single-shot accumulation is hypothesized. Moreover, the gas phase dissociation was minimized by parametric optimization [72]. Therefore, the possibility to observe native protein complexes brings an important outlook for the newly established MALDI-MS imaging that leads native proteomics directly to a tissue specimen contributing to the histopathological medical diagnosis field [73][. The “native-MSI” methodology was further improved by using liquid extraction surface analysis in order to spatially resolve and identify intact proteins with a molecular weight up to 47 kDa, including trimeric protein complex, directly on rat kidney tissue section [74].
References
- Karas, M.; Bachmann, D.; Bahr, U.; Hillenkamp, F. Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. Ion Process. 1987, 78, 53–68.
- Hillenkamp, F.; Karas, M. The MALDI Process and Method. In Maldi Ms; John Wiley & Sons: Weinheim, Germany; pp. 1–28.
- Josip, L. Introducing Proteomics: From Concepts to Sample Separation, Mass Spectrometry and Data Analysis; Wiley-Blackwell: West Sussex, UK, 2014.
- Noor, Z.; Ahn, S.B.; Baker, M.S.; Ranganathan, S.; Mohamedali, A. Mass spectrometry–based protein identification in proteomics—A review. Brief. Bioinform. 2020.
- Bolbach, G. Matrix-Assisted Laser Desorption/Ionization Analysis of Non-Covalent Complexes: Fundamentals and Applications. Curr. Pharm. Des. 2005, 11, 2535–2557.
- Kokkinidis, M.; Glykos, N.M.; Fadouloglou, V.E. Chapter 7—Protein Flexibility and Enzymatic Catalysis. In Advances in Protein Chemistry and Structural Biology; Christov, C., Karabencheva-Christova, T., Eds.; Academic Press: Cambridge, MA, USA; Volume 87, pp. 181–2018.
- Rousseau, F.; Schymkowitz, J. A systems biology perspective on protein structural dynamics and signal transduction. Curr. Opin. Struct. Biol. 2005, 15, 23–30.
- Mythily, S.; Roger, W.R. Immunomodulatory Peptides from IgSF Proteins: A Review. Curr. Protein Pept. Sci. 2005, 6, 185–196.
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; Wiley: Hoboken, NJ, USA, 2011; p. 1428 S.
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55.
- Wilson, I.A.; Stanfield, R.L. Antibody-antigen interactions: New structures and new conformational changes. Curr. Opin. Struct. Biol. 1994, 4, 857–867.
- Al Qaraghuli, M.M.; Kubiak-Ossowska, K.; Ferro, V.A.; Mulheran, P.A. Antibody-protein binding and conformational changes: Identifying allosteric signalling pathways to engineer a better effector response. Sci. Rep. 2020, 10, 13696.
- Halakou, F.; Gursoy, A.; Keskin, O. Embedding Alternative Conformations of Proteins in Protein–Protein Interaction Networks. In Protein-Protein Interaction Networks: Methods and Protocols; Canzar, S., Ringeling, F.R., Eds.; Springer US: New York, NY, USA, 2020; pp. 113–124.
- McLafferty, F. Tandem mass spectrometry. Science 1981, 214, 280–287.
- Loo, J.A. Electrospray ionization mass spectrometry: A technology for studying noncovalent macromolecular complexes. Int. J. Mass Spectrom. 2000, 200, 175–186.
- Daniel, J.M.; Friess, S.D.; Rajagopalan, S.; Wendt, S.; Zenobi, R. Quantitative determination of noncovalent binding interactions using soft ionization mass spectrometry. Int. J. Mass Spectrom. 2002, 216, 1–27.
- Chen, F.; Gülbakan, B.; Weidmann, S.; Fagerer, S.R.; Ibáñez, A.J.; Zenobi, R. Applying mass spectrometry to study non-covalent biomolecule complexes. Mass Spectrom. Rev. 2016, 35, 48–70.
- Mitra, G. Emerging Role of Mass Spectrometry-Based Structural Proteomics in Elucidating Intrinsic Disorder in Proteins. Proteomics 2020, 2000011.
- Boeri Erba, E.; Signor, L.; Petosa, C. Exploring the structure and dynamics of macromolecular complexes by native mass spectrometry. J. Proteom. 2020, 222, 103799.
- Simon, R.P.; Winter, M.; Kleiner, C.; Ries, R.; Schnapp, G.; Heimann, A.; Li, J.; Zuvela-jelaska, L.; Bretschneider, T.; Luippold, A.H.; et al. MALDI-TOF Mass Spectrometry-Based High-Throughput Screening for Inhibitors of the Cytosolic DNA Sensor cGAS. SAGE 2019, 25.
- Israr, M.Z.; Bernieh, D.; Salzano, A.; Cassambai, S.; Yazaki, Y.; Suzuki, T. Matrix-assisted laser desorption ionisation (MALDI) mass spectrometry (MS): Basics and clinical applications. Clin. Chem. Lab. Med. (Cclm) 2020, 58, 883.
- Weidmann, S.; Zenobi, R. High-Mass MALDI-MS Using Ion Conversion Dynode Detectors: Influence of the Conversion Voltage on Sensitivity and Spectral Quality. J. Am. Soc. Mass Spectrom. 2014, 25, 950–954.
- Wang, C.-C.; Lai, Y.-H.; Ou, Y.-M.; Chang, H.-T.; Wang, Y.-S. Critical factors determining the quantification capability of matrix-assisted laser desorption/ionization—time-of-flight mass spectrometry. Philos. Trans. R. Soc. A: Math. Phys. Eng. Sci. 2016, 374, 20150371.
- Pepelnjak, M.; de Souza, N.; Picotti, P. Detecting Protein–Small Molecule Interactions Using Limited Proteolysis–Mass Spectrometry (LiP-MS). Trends Biochem. Sci. 2020.
- Uversky, V.; Longhi, S. Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 3.
- Cohen, S.L.; Ferré-D’Amaré, A.R.; Burley, S.K.; Chait, B.T. Probing the solution structure of the DNA-binding protein Max by a combination of proteolysis and mass spectrometry. Protein Sci. 1995, 4, 1088–1099.
- Tomer, K.B.; Trojak, S.J.; Parker, C.E. Structural Studies of Protein-Protein Interactions Using Proteolytic Footprinting and MALDI/MS. In New Methods for the Study of Biomolecular Complexes; Ens, W., Standing, K.G., Chernushevich, I.V., Eds.; Springer: Dordrecht, The Netherlands, 1998; pp. 59–65.
- Kriwacki, R.W.; Wu, J.; Siuzdak, G.; Wright, P.E. Probing Protein/Protein Interactions with Mass Spectrometry and Isotopic Labeling: Analysis of the p21/Cdk2 Complex. JACS 1996, 118, 5320–5321.
- Kriwacki, R.W.; Wu, J.; Tennant, L.; Wright, P.E.; Siuzdak, G. Probing protein structure using biochemical and biophysical methods: Proteolysis, matrix-assisted laser desorption/ionization mass spectrometry, high-performance liquid chromatography and size-exclusion chromatography of p21Waf1/Cip1/Sdi1. J. Chromatogr. A 1997, 777, 23–30.
- Siuzdak, G. Probing viruses with mass spectrometry. J. Mass Spectrom. 1998, 33, 203–211.
- Zlotnick, A.; Reddy, V.S.; Dasgupta, R.; Schneemann, A.; Ray, W.J.; Rueckert, R.R.; Johnson, J.E. Capsid assembly in a family of animal viruses primes an autoproteolytic maturation that depends on a single aspartic acid residue. J. Biol. Chem. 1994, 269, 13680–13684.
- Zhao, Y.; Xiao, K. A Mass Spectrometry-Based Structural Assay for Activation-Dependent Conformational Changes in β-Arrestins. In Beta-Arrestins; Humana Press: New York, NY, USA, 2019; pp. 293–308.
- Xiao, K.; Shenoy, S.K.; Nobles, K.; Lefkowitz, R.J. Activation-dependent conformational changes in {beta}-arrestin 2. J. Biol. Chem. 2004, 279, 55744–55753.
- Shao, J.; Irwin, A.; Hartson, S.D.; Matts, R.L. Functional Dissection of Cdc37: Characterization of Domain Structure and Amino Acid Residues Critical for Protein Kinase Binding. Biochemistry 2003, 42, 12577–12588.
- Dhungana, S.; Williams, J.G.; Fessler, M.B.; Tomer, K.B. Epitope Mapping by Proteolysis of Antigen–Antibody Complexes. In Epitope Mapping Protocols: Second Edition; Schutkowski, M., Reineke, U., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 87–101.
- Chalmers, M.J.; Busby, S.A.; Pascal, B.D.; West, G.M.; Griffin, P.R. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein–ligand interactions. Expert Rev. Proteom. 2011, 8, 43–59.
- Chapter 2 Hydrogen Exchange. Advances in Heterocyclic Chemistry Volume 47; Katritzky, A.R., Taylor, R., Eds.; Academic Press: San Diego, CA, USA, 1990; Volume 47, pp. 7–37.
- Persson, F.; Halle, B. How amide hydrogens exchange in native proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10383–10388.
- Weis, D.D. Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications; John Wiley & Sons: West Sussex, UK, 2016.
- Englander, S.W. Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom. 2006, 17, 1481–1489.
- Beckett, D. Hydrogen–Deuterium Exchange Study of an Allosteric Energy Cycle. In Allostery: Methods and Protocols; Fenton, A.W., Ed.; Springer: New York, NY, USA, 2012; pp. 261–278.
- Hodge, E.A.; Benhaim, M.A.; Lee, K.K. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 2020, 29, 843–855.
- Mandell, J.G.; Falick, A.M.; Komives, E.A. Identification of protein–protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. USA 1998, 95, 14705–14710.
- Mandell, J.G.; Falick, A.M.; Komives, E.A. Measurement of Amide Hydrogen Exchange by MALDI-TOF Mass Spectrometry. Anal. Chem. 1998, 70, 3987–3995.
- Narayanan, S.; Mitra, G.; Muralidharan, M.; Mathew, B.; Mandal, A.K. Protein Structure–Function Correlation in Living Human Red Blood Cells Probed by Isotope Exchange-based Mass Spectrometry. Anal. Chem. 2015, 87, 11812–11818.
- Woofter, R.T.; Maurer, M.C. Role of calcium in the conformational dynamics of factor XIII activation examined by hydrogen–deuterium exchange coupled with MALDI-TOF MS. Arch. Biochem. Biophys. 2011, 512, 87–95.
- McDonald, C.; Li, L. Limited proteolysis combined with isotope labeling and quantitative LC-MALDI MS for monitoring protein conformational changes: A study on calcium-binding sites of cardiac Troponin C. Anal. Chim. Acta 2005, 534, 3–10.
- Rand, K.D.; Bache, N.; Nedertoft, M.M.; Jørgensen, T.J.D. Spatially Resolved Protein Hydrogen Exchange Measured by Matrix-Assisted Laser Desorption Ionization In-Source Decay. Anal. Chem. 2011, 83, 8859–8862.
- Pirrone, G.F.; Wang, H.; Canfield, N.; Chin, A.S.; Rhodes, T.A.; Makarov, A.A. Use of MALDI-MS Combined with Differential Hydrogen–Deuterium Exchange for Semiautomated Protein Global Conformational Screening. Anal. Chem. 2017, 89, 8351–8357.
- Alexovič, M.; Urban, P.L.; Tabani, H.; Sabo, J. Recent advances in robotic protein sample preparation for clinical analysis and other biomedical applications. Clin. Chim. Acta 2020, 507, 104–116.
- Kabaria, S.R.; Mangion, I.; Makarov, A.A.; Pirrone, G.F. Use of MALDI-MS with solid-state hydrogen deuterium exchange for semi-automated assessment of peptide and protein physical stability in lyophilized solids. Anal. Chim. Acta 2019, 1054, 114–121.
- Reddy, N.C.; Kumar, M.; Molla, R.; Rai, V. Chemical methods for modification of proteins. Org. Biomol. Chem. 2020, 18, 4669–4691.
- Zhang, A.; Hu, P.; MacGregor, P.; Xue, Y.; Fan, H.; Suchecki, P.; Olszewski, L.; Liu, A. Understanding the Conformational Impact of Chemical Modifications on Monoclonal Antibodies with Diverse Sequence Variation Using Hydrogen/Deuterium Exchange Mass Spectrometry and Structural Modeling. Anal. Chem. 2014, 86, 3468–3475.
- Chen, F.; Gerber, S.; Heuser, K.; Korkhov, V.M.; Lizak, C.; Mireku, S.; Locher, K.P.; Zenobi, R. High-Mass Matrix-Assisted Laser Desorption Ionization-Mass Spectrometry of Integral Membrane Proteins and Their Complexes. Anal. Chem. 2013, 85, 3483–3488.
- Chavez, J.D.; Mohr, J.P.; Mathay, M.; Zhong, X.; Keller, A.; Bruce, J.E. Systems structural biology measurements by in vivo cross-linking with mass spectrometry. Nat. Protoc. 2019, 14, 2318–2343.
- Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol. 2020, 63, 48–53.
- Chang, Z.; Kuchar, J.; Hausinger, R.P. Chemical cross-linking and mass spectrometric identification of sites of interaction for UreD, UreF, and urease. J. Biol. Chem. 2004, 279, 15305–15313.
- Nguyen-Huynh, N.-T.; Sharov, G.; Potel, C.; Fichter, P.; Trowitzsch, S.; Berger, I.; Lamour, V.; Schultz, P.; Potier, N.; Leize-Wagner, E. Chemical cross-linking and mass spectrometry to determine the subunit interaction network in a recombinant human SAGA HAT subcomplex. Protein Sci. 2015, 24, 1232–1246.
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: San Diego, CA, USA, 2013. [Google Scholar]
- Wong, S.S. Chemistry of Protein Conjugation and Cross-Linking; CRC Press: Boca Raton, FL, USA, 1991.
- Müller, M.Q.; Zeiser, J.J.; Dreiocker, F.; Pich, A.; Schäfer, M.; Sinz, A. A universal matrix-assisted laser desorption/ionization cleavable cross-linker for protein structure analysis. Rapid Commun. Mass Spectrom. 2011, 25, 155–161.
- Mädler, S.; Barylyuk, K.; Boeri Erba, E.; Nieckarz, R.J.; Zenobi, R. Compelling Advantages of Negative Ion Mode Detection in High-Mass MALDI-MS for Homomeric Protein Complexes. J. Am. Soc. Mass Spectrom. 2012, 23, 213–224.
- Köhler, M.; Neff, C.; Perez, C.; Brunner, C.; Pardon, E.; Steyaert, J.; Schneider, G.; Locher, K.P.; Zenobi, R. Binding Specificities of Nanobody•Membrane Protein Complexes Obtained from Chemical Cross-Linking and High-Mass MALDI Mass Spectrometry. Anal. Chem. 2018, 90, 5306–5313.
- Krauth, F.; Ihling, C.H.; Rüttinger, H.H.; Sinz, A. Heterobifunctional isotope-labeled amine-reactive photo-cross-linker for structural investigation of proteins by matrix-assisted laser desorption/ionization tandem time-of-flight and electrospray ionization LTQ-Orbitrap mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 2811–2818.
- Sinz, A.; Kalkhof, S.; Ihling, C. Mapping protein interfaces by a trifunctional cross-linker combined with MALDI-TOF and ESI-FTICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2005, 16, 1921–1931.
- Ptáčková, R.; Ječmen, T.; Novák, P.; Hudeček, J.; Stiborová, M.; Šulc, M. The application of an emerging technique for protein-protein interaction interface mapping: The combination of photo-initiated cross-linking protein nanoprobes with mass spectrometry. Int. J. Mol. Sci. 2014, 15, 9224–9241.
- Li, G.; Ma, F.; Cao, Q.; Zheng, Z.; DeLaney, K.; Liu, R.; Li, L. Nanosecond photochemically promoted click chemistry for enhanced neuropeptide visualization and rapid protein labeling. Nat. Commun. 2019, 10, 4697.
- Petrotchenko, E.V.; Xiao, K.; Cable, J.; Chen, Y.; Dokholyan, N.V.; Borchers, C.H. BiPS, a Photocleavable, Isotopically Coded, Fluorescent Cross-linker for Structural Proteomics. Mol. Cell. Proteom. 2009, 8, 273–286.
- Rosinke, B.; Strupat, K.; Hillenkamp, F.; Rosenbusch, J.; Dencher, N.; Krüger, U.; Galla, H.-J. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of membrane proteins and non-covalent complexes. J. Mass Spectrom. 1995, 30, 1462–1468.
- Cohen, S.L.; Padovan, J.C.; Chait, B.T. Mass Spectrometric Analysis of Mercury Incorporation into Proteins for X-ray Diffraction Phase Determination. Anal. Chem. 2000, 72, 574–579.
- Wortmann, A.; Pimenova, T.; Alves, S.; Zenobi, R. Investigation of the first shot phenomenon in MALDI mass spectrometry of protein complexes. Analyst 2007, 132, 199–207.
- Beaufour, M.; Ginguené, D.; Le Meur, R.; Castaing, B.; Cadene, M. Liquid Native MALDI Mass Spectrometry for the Detection of Protein-Protein Complexes. J. Am. Soc. Mass Spectrom. 2018, 29, 1981–1994.
- Griffiths, R.L.; Cooper, H.J. Direct Tissue Profiling of Protein Complexes: Toward Native Mass Spectrometry Imaging. Anal. Chem. 2016, 88, 606–609.
- Hale, O.J.; Cooper, H.J. Native Mass Spectrometry Imaging and In Situ Top-Down Identification of Intact Proteins Directly from Tissue. J. Am. Soc. Mass Spectrom. 2020.

