Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Valentine Chapelle | -- | 3720 | 2022-10-18 14:42:11 | | | |
| 2 | Rita Xu | -22 word(s) | 3698 | 2022-10-19 05:05:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chapelle, V.; Silvestre, F. DNA Methylation Variation in Wild Animal Populations. Encyclopedia. Available online: https://encyclopedia.pub/entry/29957 (accessed on 27 June 2026).
Chapelle V, Silvestre F. DNA Methylation Variation in Wild Animal Populations. Encyclopedia. Available at: https://encyclopedia.pub/entry/29957. Accessed June 27, 2026.
Chapelle, Valentine, Frédéric Silvestre. "DNA Methylation Variation in Wild Animal Populations" Encyclopedia, https://encyclopedia.pub/entry/29957 (accessed June 27, 2026).
Chapelle, V., & Silvestre, F. (2022, October 18). DNA Methylation Variation in Wild Animal Populations. In Encyclopedia. https://encyclopedia.pub/entry/29957
Chapelle, Valentine and Frédéric Silvestre. "DNA Methylation Variation in Wild Animal Populations." Encyclopedia. Web. 18 October, 2022.
Copy Citation
Population epigenetics explores the extent of epigenetic variation and its dynamics in natural populations encountering changing environmental conditions. In contrast to population genetics, the basic concepts of this field are still in their early stages, especially in animal populations. Epigenetic variation may play a crucial role in phenotypic plasticity and local adaptation as it can be affected by the environment, it is likely to have higher spontaneous mutation rate than nucleotide sequences do, and it may be inherited via non-mendelian processes.
population epigenetics
DNA methylation variation
epimutation
1. Introduction
The understanding of an organism’s capacity to respond to environmental changes has advanced, in a large way, through studies focusing on genetic variation and the manipulation of environmental conditions. These studies confirm that genotype, environment, and their interaction contribute to phenotypic variability, a fundamental prerequisite for evolution by natural selection. The tremendous development of genetic knowledge during the 20th century has led to the merge of Darwinism and the field of genetics into a modern synthesis. However, researchers now admit that genetic variation is not the only source of phenotypic variation that can be inherited across generations because only a small proportion of variance in complex traits can actually be explained by genetic variance [1]. The concept of inclusive heritability has been proposed to unify genetic and non-genetic mechanisms of heritability, which encompasses all dimensions of inheritance such as the transmitted parental effect, ecological variation, social variation, and transgenerational epigenetic inheritance (TEI) [2]. There is a growing consensus that epigenetics and, in particular, TEI could be one of the missing factors for understanding phenomena that cannot be explained by the DNA sequence alone, such as incomplete penetrance (i.e., individuals of a given genotype expressing different phenotypes) and the variance in expressivity (i.e., the degree/intensity to which complex trait expression differs among individuals) [3][4]. These two phenomena result in an incomplete correlation between genotype and phenotype, and these may be partly explained by epigenetic mechanisms.
Epigenetics has been narrowly defined as mitotically and/or meiotically heritable changes in gene expression that cannot be explained by changes in the gene sequence [5]. These changes include histone modification, DNA methylation, and small RNA regulation, and these are involved in processes such as cellular differentiation, development, diseases, behaviors, and metabolism [6]. Studies exploring phenotypic plasticity have showed that epigenetic variation can play a significant role in the response that an organism has to environmental variation, as epigenetic marks can be directly affected by the environment. In other words, environmentally induced epigenetic variations have been proposed to mediate phenotypic plasticity as they allow the organisms to adapt to the environmental conditions by increasing the phenotypic options of a genotype with no genetic sequence modification [7][8][9]. Moreover, fitness-related phenotypes that are initially environmentally induced can be selected to become genetically determined, and hence, heritable, a process that is named genetic accommodation [10][11]. Genetic assimilation (i.e., the loss of or decreased plasticity [12]) and genetic compensation (i.e., the selection for similar phenotypes in different environments [13]) are different types of genetic accommodation. In other words, in addition to being another source of phenotypic variation, epigenetic variation can precede genetic adaptation through genetic accommodation, thus reversing the standard model of evolution from a genotype-to-phenotype to a phenotype-to-genotype information flow [11][14][15].
Understanding the evolutionary implications of epigenetics and how epigenetic mechanisms contribute to phenotypic variability is one of the current greatest challenges in evolutionary biology. The importance of epigenetic variation in environmental adaptation and evolution has been investigated much more in plants than it has been in animals [16][17]. Moreover, most epigenetic data that are available on animals have been collected under laboratory conditions in model organisms such as mice and insects and these studies have mainly focused on epigenetic mechanisms and their responses to environmental stressors [17][18][19][20]. Laboratory studies on plants and animals have shed light on some of the general features of epigenetics, with important evolutionary implications. First, epimutations were assessed to be up to five orders of magnitude more frequent than genetic mutations were (10−4 versus 10−9 per base pair and generation) in the model plant Arabidopsis thaliana [21][22]. Second, some epigenetic marks may be stably inherited across generations through transgenerational epigenetic inheritance, as reported in many plant and animal taxa such as mammals [23], birds [24], fish [25], and invertebrates [26][27]. Third, epigenetic variation that is associated with changes in gene expression can be environmentally induced in plants and animals [28][29].
An important step is now to examine the extent of epigenetic variation and the way that this variation changes over time in wild populations that encounter natural levels of environmental complexity, genetic structure and dynamics, and natural ecological processes. This endeavor represents part of the field of population epigenetics. Though the basic concepts of population genetics from the 1930s are well described, these have been extended with the introduction of the modern synthesis (MS), however, the body of knowledge concerning population epigenetics remains largely scarce as it is a new research interest. As for laboratory studies, most of the natural population, epigenetic research projects have been carried out in plant populations, which have been reviewed elsewhere [16][30][31]. The first experimental work investigating epigenetics in natural animal populations was published in 2010 on DNA methylation in rainbow trout Oncorhynchus mykiss [32]. There has been a growing number of studies focusing on natural population epigenetic variation, especially DNA methylation variation in animals in terms of their phenotypic diversity generation and local adaptation.
DNA methylation, which is the most extensively characterized epigenetic mechanism in both plants and animals [33]. DNA methylation is found across all taxa of life, and primarily occurs at the 5-methylcytosine bases in eukaryotes and prokaryotes [34]. It involves the addition of a methyl group to cytosine within the CpG dinucleotides in animals. The DNA methylation of regulatory regions is generally associated with gene down-regulation or silencing, but that is not always the case [35][36]. Recent studies have showed that gene body methylation is positively correlated with transcriptional activity in most animal species [33]. The genomic distribution of DNA methylation has been described in many clades of animals, but there are some differences in how and where it occurs. In vertebrates, the pattern and extent of DNA methylation is well conserved across species; DNA methylation occurs nearly throughout the entire genome, with 70–80% of cytosines in the CpG dinucleotides being methylated [37]. Gene bodies, including exons and introns, are typically methylated, while CpGs in the gene promoter regions are often lowly methylated [38][39]. The idea that only vertebrates have a highly methylated genome has recently been challenged as this phenomena has also been found in the sponge Amphimedon queenslandica and a unicellular green algae from the genus Chlorella [34][40]. Despite this rather consistent DNA methylation pattern across vertebrate species, differences occur in terms of the pattern establishment during early embryogenesis. Taking DNA methylation reprogramming as an example, the demethylation of both parental genomes occurs in the mouse embryo, whereas the paternal pattern of methylation is maintained in zebrafish, with a reprogramming of the maternal DNA to correspond to the paternal template [41]. Regarding invertebrates, DNA methylation patterns are extremely variable across taxa. Some invertebrate genomes lack cytosine methylation such as the nematode Caenorhabditis elegans, the platyhelminth Schmidtea mediterranea, the fruit fly Drosophila melanogaster, and the rotifer Adineta vaga [42][43], while others are similar to plants as they have a mosaic of heavily methylated DNA domains (predominantly in exons) that are interspersed with domains that are methylation-free, such as the sea anemone, honey bee, and sea squirt [34][44]. Although the DNA methylation pattern and its genomic distribution vary widely across animal taxa, it is possible to draw general lines on its diversity and its responsiveness when it is facing natural environmental conditions.
2. Epigenetic Diversity in Natural Animal Populations
DNA sequences are a succession of four different bases (A, C, T, and G), and each mutation switches a base for another one. An allele is a variant of the same gene that is located at the same genetic locus and is characterized by a specific sequence. Each diploid organism owns two alleles at each locus, and it is qualified as heterozygous if the alleles are different, or homozygous if they are the same. The situation is quite different for DNA methylation marks, since a cytosine can only be methylated (M) or unmethylated (U), thus restricting to two the number of possibilities that there can be at each cytosine. For the same allele, each CpG (and at a lower level, each CHG and CHH-H for any base, except for G) can either be M or U, thus producing a succession of single methylation polymorphisms (SMPs). SMPs can accumulate in the DNA sequence, and they produce a specific methylation pattern, or epiallele. While genetic variation refers to the different allele frequencies that there are among individuals or populations, epigenetic variation corresponds to the presence or absence of epigenetic markers at specific loci that are studied among and/or within populations [45]. The amount of epigenetic variation within a population is called epigenetic diversity, and it refers to SMP diversity [46]. SMP diversity is generated by epimutations, i.e., epigenetic modifications at a given position or region, and its origins can be genetic, environmental, or stochastic [45][47]. Focusing on cytosine methylation (5mC), epimutations are heritable changes in the methylation status of a single cytosine or of a region or cluster of cytosines [48]. To determine the DNA methylation diversity in wild animal populations, most field studies have used methylation-sensitive amplified polymorphism (MSAP or MS-AFLP) [49][50][51]. Next-generation sequencing is, more rarely and most recently, the method that has been used among other such as reduced representation bisulfite sequencing (RRBS) [52][53][54][55], MeDIP-Seq analysis [56][57], and whole-genome bisulfite sequencing (WGBS) [58].
By comparing the epigenetic and genetic diversity in wild animal populations, it is possible to estimate the relative importance of genetic and epigenetic variation in populations phenotypic diversity, and to test the hypothesis that epigenetic divergence acts as the first step in speciation, allowing for the expression of alternative phenotypes in response to environmental changes, which are ultimately fixed by genetic accommodation or assimilation [11][15][59][60]. Many studies have identified extensive epigenetic diversity that exceeds the genetic diversity between natural populations of plants [61][62]. It was suggested that the epigenetic variation in natural plant populations plays a major role for their transient and/or heritable adjustment to the changing environments, as it may be stable and related to environmental variation [63]. This implies that these environmentally induced epimutations may lead to the convergence of individuals that are living in similar habitat conditions; this is a situation that may be exacerbated by TEI. Despite the fact that studies on epigenetic variation in natural animal populations are scarce when they are compared to plant studies, some discernible patterns have emerged after researchers have reviewed them. Regardless of whether researchers focused on crustaceans, mollusks, fish, reptiles, birds, or mammals, the DNA methylation variation was larger than the genetic variation was among and/or within wild animal populations [53][56][64][65][66][67][68]. For example, Smith et al. studied the DNA methylation variation in fish (Etheostoma olmstedi) using the MSAP technique. They investigated two North American river drainages, wherein, each of them include several closely related populations, to characterize the epigenetic variation within and among the populations. They obtained results that demonstrated that there is a significantly greater epigenetic diversity than there is genetic diversity within all of the populations in both the Patuxent and Potomac rivers. Regarding the diversity among the populations, their analysis demonstrated that there is a substantial epigenetic structure, but no genetic structure, meaning that E. olmstedi populations are significantly different from each other in terms of their DNA methylation patterns, but not in terms of their genomes [60]. They assumed that the methylome is changing faster than the genome is in this species, which is in accordance with the general hypothesis that epigenetic divergence can precede genetic divergence in evolution due to its dynamics.
A larger amount of epigenetic diversity in comparison to the amount of genetic diversity can also arise between populations of sister species. Skinner et al. measured the genetic mutations (via copy number variation—CNV) and epimutations (via differential DNA methylation regions—DMRs) across five species of Darwin’s finches (Geospiza fuliginosa, G. fortis, G. scandens, Camarhynchus parvulus, and Platyspiza crassirostris). As a result of these measurements, they found that there were fewer genetic mutations than there were epimutations among the five species, showing that the differences in the methylome are more related to evolutionary relationships than they are differences in the genome. Moreover, they reported that the differentially methylated genes were related to evolutionarily important pathways in birds [65]. Vernaz et al. found a substantial methylome divergence between six Lake Malawi cichlid species that show extensive phenotypic diversity despite having them extremely low DNA sequence divergence. These DMRs were enriched in transposons and were associated with the transcription changes of ecologically relevant genes that are related to energy expenditure and lipid metabolism in the cichlid’s liver [68].
An extreme situation can be observed in asexual species exhibiting a lack of genetic variation due to their reproductive system. A study on epigenetic polymorphism in the clonal fish Chrosomus eos-neogaeus from seven geographically distant lakes showed that they have an interindividual DNA methylation variability. Moreover, individuals could be regrouped according to their lake of origin on the basis of their unique methylation profile, as individuals of a given lake are epigenetically similar [69][70]. Thorson et al. measured the genome-wide DNA methylation variation of asexual New Zealand freshwater snail Potamopyrgus antipodarum from distinct habitats (two lakes versus two rivers). Those snails have significant methylation signatures when one is comparing those of the lake versus those of the river populations [71]. Later, they examined the methylation variation among those in the lakes that differ in their environmental disturbance and pollution histories. Using an MeDIP-Seq analysis, they showed the presence of site-specific differences in the DNA methylation between each of those lake populations [57]. These studies raise the question of the environmental implications in epigenetic variability, which is discussed later.
In most of the wild animal populations that have been examined to date, independently of the studied organisms or the molecular analysis that is being used, the DNA methylation variation is larger than the variation in allele frequencies within and among natural animal populations. This should not be surprising as epimutations can happen randomly, such as mutations, but they can also be triggered by environmental conditions and by the genotype itself. The epigenetic diversity that is found in a population is therefore the result of the combination of these three distinct sources. To determine the implications of the epigenetic processes in evolution, a major concern is to characterize the degree of autonomy between epigenetic and genetic variation and ultimately, the degree of phenotypic variation that can be explained only by the environmental or stochastic epimutations [72].
3. Correlation between Epigenetic and Genetic Variation in Natural Animal Populations
Based on their degree of autonomy from the underlying genotype, epialleles are categorized into three types: obligatory, which is completely dependent on the genetic variation; facilitated, which is directed or loosely potentiated by the genotype; pure, which is independent of the genetic variation and is generated by stochastic events or environmental changes [73]. To identify which epialleles categories are encountered in natural populations, the correlation between the genetic and epigenetic profiles can be estimated, mostly by using a Mantel test [50][63][74][75]. A significant (positive or negative) correlation suggests that the epigenetic and genetic variations are interdependent, which corresponds to the presence of obligatory epialleles. In contrast, the absence of a significant correlation suggests that the epigenetic variation can autonomously impact the phenotypic variation, by being totally or partly independent from genetic control.
Under laboratory conditions, epimutations are expected to be mostly obligatory. The lack of environmental fluctuations in the laboratory housing conditions does not promote environmentally induced epimutations and the selection of epimutation-sensitive alleles that are responsible for alternative phenotypes that occur while experiencing environmental changes in natural conditions (see the Baldwin effect in “Section 5.2 Epigenetics and macroevolution of natural animal population”). In this case, epigenetic variation can be viewed as a phenotypic read-out downstream of the genotype, with a low environmental contribution. For example, Hu and Barrett reviewed the epigenetically encoded thermal plasticity in animals. Of the 14 studies, 13 included a putatively obligatory epigenetic variation that was underlying phenotypic plasticity, and only one was categorized as “unknown” [45]. In mice and humans, some studies have evaluated the association between epigenetic and genetic variation with a narrow-sense heritability, i.e., the ratio of additive genetic variance to the total phenotypic variance. It appears that genetic variation can explain an average of 7-34 % of all methylation variation [76][77][78]. In natural animal populations, although several studies have measured epigenetic and genetic variation, only a few of them have estimated the relationship between those variations. Of the 26 reviewed studies, 12 did not calculate a correlation coefficient between the genetic and epigenetic variation, eight studies found a non-significant correlation, and six studies obtained a significant correlation (Table 1). Moreover, few authors have linked a calculated correlation coefficient to Richards’ epiallele categories. For instance, Liebl et al. obtained a significant negative correlation between the genetic and DNA methylation variation within seven populations of house sparrows (Passer domesticus) [79]. However, they predicted that all three kinds of epialleles could play a role in those populations as their design could not discriminate between the three categories. Despite the lack of direct connection between these categories and the genetic vs. epigenetic variation–correlation coefficient, calculating this coefficient can still help to estimate the relative importance of the genetic and epigenetic variation in the mechanisms that are facilitating population divergence, and to highlight the extent to which epigenetic variation is under genetic control [67][73].
Table 1. Overview of studies focusing on genetic and epigenetic diversity and correlation in natural animal populations.
| Species | Genetic vs. Epigenetic Correlation | Epialleles Category | Ref. |
|---|---|---|---|
| Clonal fish (Chrosomus eos-neogaeus) |
No significant correlation | Putatively pure or facilitated | [69] |
| Clonal fish (Chrosomus eos-neogaeus) |
No significant correlation | Putatively pure or facilitated | [70] |
| Clonal fish (Chrosomus eos-neogaeus) |
No significant correlation | Unknown | [80] |
| House sparrows (Passer domesticus) (Africa) |
Significant negative correlation | Unknown | [79] |
| House sparrows (Passer domesticus) (Australia) |
No significant correlation | Unknown | [74] |
| Red grouse (Lagopus lagopus scotica) |
No significant correlation | Unknown | [81] |
| Bats (Rhinolophus pusillus, Hipposideros armiger and Miniopterus fuliginosus) |
Significant positive correlation | Unknown | [66] |
| South African (Gansbaii) sandhopper (Talorchestia capensis) |
No significant correlation | Putatively pure or facilitated | [50] |
| South African sandhopper (Talorchestia capensis) |
Significant negative correlation | Putatively obligatory | [50] |
| Pacific oyster (Crassostrea gigas) |
Significant positive correlation | Putatively obligatory | [67] |
| Crested anole (Anolis cristatellus) |
Significant positive correlation | Putatively obligatory | [54] |
| Eastern oyster (Crassostrea virginica) |
No significant correlation | Unknown | [53] |
| Fish (Gobio occitaniae) |
Significant positive correlation | Unknown | [75] |
| Chinook salmon (Oncorhynchus tshawytscha) |
No significant correlation | Unknown | [82] |
Despite the fact that the obligatory epialleles’ relevance is questioned regarding its evolutionary potential [69], an interesting new insight is that the epigenome can also influence the genome, thereby creating a significant relationship between their variation. Firstly, the epigenetic variation can regulate the active status of transposable elements (TEs). TEs are DNA sequences that have the ability to change their position within a genome. The epigenetic control of gene expression mostly originates from the regulation of TEs that are inserted near genes [83]. In fact, TEs are the major carriers of epigenetic marks and are subject to almost all epigenetic regulatory mechanisms in plants [84]. Interestingly, there is a great variability in the locations of the TEs, not only between different species, but also within populations. More than 90% of the TEs that are inserted at a specific genomic position are not present in all individuals within both animal [85] and plant [84] populations. Transposon insertion polymorphism between individuals may come from epigenetic variation and it results in genomic sequence variation. Secondly, DNA methylation variation can influence the mutation rates in repetitive elements which are known to regulate genome 3D folding and the establishment of heterochromatin, among other regulation mechanisms [86]. These repetitive elements are patterns of nucleic acids that occur as multiple copies in the DNA sequence, comprising TEs, simple sequence repeats (SSRs), and microsatellites, and these represent a major fraction of vertebrate genomes [87]. Some studies have highlighted the correlation between a decrease in the DNA methylation of specific repetitive elements and an increase in the copy number variations (CNVs), thus showing another possible epigenetic-to-genetic flow [88][89]. Finally, besides these specific genome components, epimutations may alter the global genome stability and modify the mutation rate of the DNA sequences throughout the genome. Methylated cytosines (mCs) in the CpG context seem to have a higher mutation rate than non-methylated ones [90], and this is in part because mCs are subject to spontaneous deamination [91]. This process, where an mC turns into a T, occurs at a rate that is 10- to 50-fold higher than any other mutation is in humans [92]. The result of this hypermutability is a CpG depletion in the consistently methylated genomic regions. Yet, the amount of CpGs in a genome partly determines its epigenetic potential, which is defined as “the capacity for environmentally induced phenotypic change (i.e., plasticity) via epigenetic modifications to relevant genomic elements” [93]. CpGs are considered as the capacitors of phenotypic plasticity; the more CpGs an organism has, then the more facilitated their acclimation is via DNA methylation and gene expression regulation.
In summary, epigenetic and genetic changes most likely work in concert to regulate the gene expression and phenotypic variation of complex traits (Figure 1). The proportion of the genotype-independent and -dependent epigenetic variation reflects the underlying mechanisms of the natural animal population’s evolutionary pathways to promote phenotypic variation [65]. New insights that researchers would like to highlight is that even though there is a significant correlation between epigenetic and genetic variation, epigenetic variation is not necessarily dependent on genetic variation. As has already been explained, the epigenome can influence the genome in different ways. Moreover, a significant correlation does not imply that there is a causal relationship. Geographical and ecological processes may create parallel evolutions of genetic and epigenetic structures, and thus, similar patterns that are without any functional link with each other. To better understand these dynamics, it is important to consider the mechanisms that are influencing both genetic and epigenetic diversity, and the processes that only act on epigenetic markers in wild animal populations.
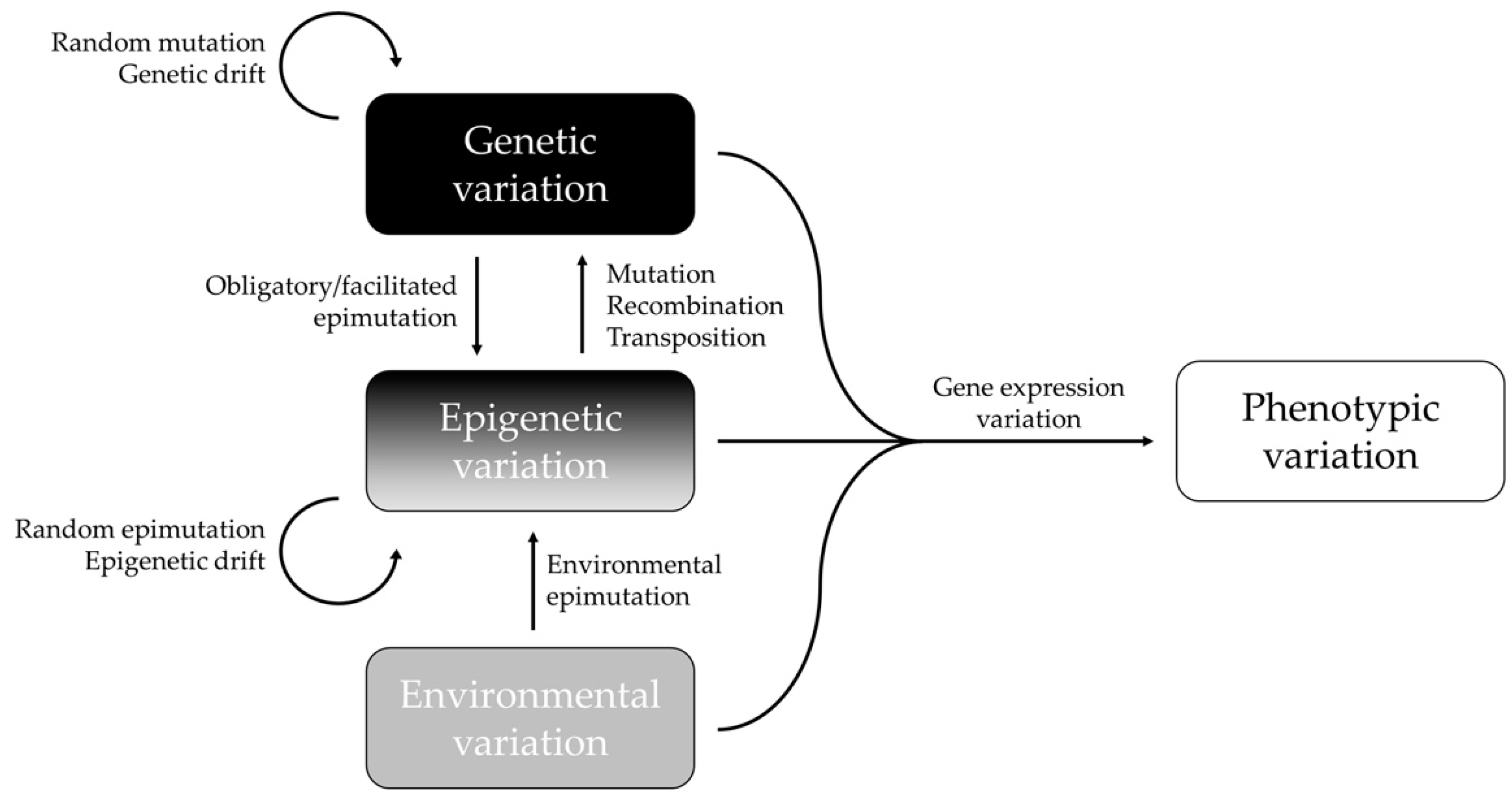
Figure 1. Interactions between epigenetic, genetic, environmental, and phenotypic variations. Epigenetic variation can depend upon the genotype (obligatory and facilitated epimutations), or it can be independent of the genotype (pure epimutations) and be generated by environmental changes or stochastic events (random epimutation/epigenetic drift). Adapted with permission from [47], 2022, Frédéric Silvestre.
References
- Mameli, M. Nongenetic Selection and Nongenetic Inheritance. Br. J. Philos. Sci. 2004, 55, 35–71.
- Danchin, É.; Charmantier, A.; Champagne, F.A.; Mesoudi, A.; Pujol, B.; Blanchet, S. Beyond DNA: Integrating Inclusive Inheritance into an Extended Theory of Evolution. Nat. Rev. Genet. 2011, 12, 475–486.
- Allen, N.D.; Norris, M.L.; Surani, M.A. Epigenetic Control of Transgene Expression and Imprinting by Genotype-Specific Modifiers. Cell 1990, 61, 853–861.
- Miko, I. Phenotype Variability: Penetrance and Expressivity. Nat. Educ. 2008, 1, 137.
- Youngson, N.A.; Whitelaw, E. Transgenerational Epigenetic Effects. Annu. Rev. Genom. Hum. Genet. 2008, 9, 233–257.
- Nicoglou, A.; Merlin, F. Epigenetics: A Way to Bridge the Gap between Biological Fields. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2017, 66, 73–82.
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally Induced Phenotypes and DNA Methylation: How to Deal with Unpredictable Conditions until the next Generation and After. Mol. Ecol. 2010, 19, 1283–1295.
- Greally, J.M. Population Epigenetics. Curr. Opin. Syst. Biol. 2017, 1, 84–89.
- Verhoeven, K.J.F.; Preite, V. Epigenetic Variation in Asexually Reproducing Organisms. Evolution 2014, 68, 644–655.
- West-Eberhard, M.J. Developmental Plasticity and Evolution; Oxford University Press: New York, NY, USA, 2003; ISBN 978-0-19-802856-7.
- West-Eberhard, M.J. Developmental Plasticity and the Origin of Species Differences. Proc. Natl. Acad. Sci. USA 2005, 102, 6543–6549.
- Robinson, B.W.; Dukas, R. The Influence of Phenotypic Modifications on Evolution: The Baldwin Effect and Modern Perspectives. Oikos 1999, 85, 582.
- Grether, G.F. Environmental Change, Phenotypic Plasticity, and Genetic Compensation. Am. Nat. 2005, 166, E115–E123.
- Jablonka, E.; Lamb, M.J. The Inheritance of Acquired Epigenetic Variations. J. Theor. Biol. 1989, 139, 69–83.
- West-Eberhard, M.J. Alternative Adaptations, Speciation, and Phylogeny (A Review). Proc. Natl. Acad. Sci. USA 1986, 83, 1388–1392.
- Richards, E.J. Natural Epigenetic Variation in Plant Species: A View from the Field. Curr. Opin. Plant Biol. 2011, 14, 204–209.
- Vogt, G. Epigenetic Variation in Animal Populations: Sources, Extent, Phenotypic Implications, and Ecological and Evolutionary Relevance. J. Biosci. 2021, 46, 24.
- Burggren, W. Epigenetic Inheritance and Its Role in Evolutionary Biology: Re-Evaluation and New Perspectives. Biology 2016, 5, 24.
- Kumar, D.; Thakur, M.K. Effect of Perinatal Exposure to Bisphenol-A on DNA Methylation and Histone Acetylation in Cerebral Cortex and Hippocampus of Postnatal Male Mice. J. Toxicol. Sci. 2017, 42, 281–289.
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Maternal E-Cigarette Exposure Results in Cognitive and Epigenetic Alterations in Offspring in a Mouse Model. Chem. Res. Toxicol. 2018, 31, 601–611.
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational Epigenetic Instability Is a Source of Novel Methylation Variants. Science 2011, 334, 369–373.
- van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, Spectrum, and Evolutionary Dynamics of Spontaneous Epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681.
- Manikkam, M.; Guerrero-Bosagna, C.; Tracey, R.; Haque, M.M.; Skinner, M.K. Transgenerational Actions of Environmental Compounds on Reproductive Disease and Identification of Epigenetic Biomarkers of Ancestral Exposures. PLoS ONE 2012, 7, e31901.
- Guerrero-Bosagna, C.; Morisson, M.; Liaubet, L.; Rodenburg, T.B.; de Haas, E.N.; Košťál, Ľ.; Pitel, F. Transgenerational Epigenetic Inheritance in Birds. Environ. Epigenetics 2018, 4, dvy008.
- Bhandari, R.K.; vom Saal, F.S.; Tillitt, D.E. Transgenerational Effects from Early Developmental Exposures to Bisphenol A or 17α-Ethinylestradiol in Medaka, Oryzias Latipes. Sci. Rep. 2015, 5, 9303.
- Seong, K.-H.; Li, D.; Shimizu, H.; Nakamura, R.; Ishii, S. Inheritance of Stress-Induced, ATF-2-Dependent Epigenetic Change. Cell 2011, 145, 1049–1061.
- Liew, Y.J.; Howells, E.J.; Wang, X.; Michell, C.T.; Burt, J.A.; Idaghdour, Y.; Aranda, M. Intergenerational Epigenetic Inheritance in Reef-Building Corals. Nat. Clim. Change 2020, 10, 254–259.
- Feil, R.; Fraga, M.F. Epigenetics and the Environment: Emerging Patterns and Implications. Nat. Rev. Genet. 2012, 13, 97–109.
- Casier, K.; Boivin, A.; Carré, C.; Teysset, L. Environmentally-Induced Transgenerational Epigenetic Inheritance: Implication of PIWI Interacting RNAs. Cells 2019, 8, 1108.
- Miryeganeh, M.; Saze, H. Epigenetic Inheritance and Plant Evolution. Popul. Ecol. 2019, 62, 17–27.
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A Role for Epigenetic Regulation in the Adaptation and Stress Responses of Non-Model Plants. Front. Plant Sci. 2019, 10, 246.
- Blouin, M.S.; Thuillier, V.; Cooper, B.; Amarasinghe, V.; Cluzel, L.; Araki, H.; Grunau, C. No Evidence for Large Differences in Genomic Methylation between Wild and Hatchery Steelhead (Oncorhynchus Mykiss). Can. J. Fish. Aquat. Sci. 2010, 67, 217–224.
- de Mendoza, A.; Lister, R.; Bogdanovic, O. Evolution of DNA Methylome Diversity in Eukaryotes. J. Mol. Biol. 2020, 432, 1687–1705.
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919.
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA Hypermethylation Associated with Upregulated Gene Expression in Prostate Cancer Demonstrates the Diversity of Epigenetic Regulation. BMC Med. Genomics 2020, 13, 6.
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inf. 2019, 18, 117693511982877.
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and Divergence of Methylation Patterning in Plants and Animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694.
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476.
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic Memory at Embryonic Enhancers Identified in DNA Methylation Maps from Adult Mouse Tissues. Nat. Genet. 2013, 45, 1198–1206.
- de Mendoza, A.; Hatleberg, W.L.; Pang, K.; Leininger, S.; Bogdanovic, O.; Pflueger, J.; Buckberry, S.; Technau, U.; Hejnol, A.; Adamska, M.; et al. Convergent Evolution of a Vertebrate-like Methylome in a Marine Sponge. Nat. Ecol. Evol. 2019, 3, 1464–1473.
- Potok, M.E.; Nix, D.A.; Parnell, T.J.; Cairns, B.R. Reprogramming the Maternal Zebrafish Genome after Fertilization to Match the Paternal Methylation Pattern. Cell 2013, 153, 759–772.
- Rae, P.M.M.; Steele, R.E. Absence of Cytosine Methylation at C-C-G-G and G-C-G-C Sites in the RDNA Coding Regions and Intervening Sequences of Drosophila and the RDNA of Other Higher Insects. Nucl. Acids. Res. 1979, 6, 2987–2995.
- Simpson, V.J.; Johnson, T.E.; Hammen, R.F. Caenorhabditis Elegans DNA Does Not Contain 5-Methylcytosine at Any Time during Development or Aging. Nucl. Acids. Res. 1986, 14, 6711–6719.
- Xu, X.; Li, G.; Li, C.; Zhang, J.; Wang, Q.; Simmons, D.K.; Chen, X.; Wijesena, N.; Zhu, W.; Wang, Z.; et al. Evolutionary Transition between Invertebrates and Vertebrates via Methylation Reprogramming in Embryogenesis. Natl. Sci. Rev. 2019, 6, 993–1003.
- Hu, J.; Barrett, R.D.H. Epigenetics in Natural Animal Populations. J. Evol. Biol. 2017, 30, 1612–1632.
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of Population Epigenomic Diversity. Nature 2013, 495, 193–198.
- Biwer, C.; Kawam, B.; Chapelle, V.; Silvestre, F. The Role of Stochasticity in the Origin of Epigenetic Variation in Animal Populations. Integr. Comp. Biol. 2020, 60, 1544–1557.
- Oey, H.; Whitelaw, E. On the Meaning of the Word ‘Epimutation’. Trends Genet. 2014, 30, 519–520.
- Ardura, A.; Zaiko, A.; Morán, P.; Planes, S.; Garcia-Vazquez, E. Epigenetic Signatures of Invasive Status in Populations of Marine Invertebrates. Sci. Rep. 2017, 7, 42193.
- Baldanzi, S.; Watson, R.; McQuaid, C.D.; Gouws, G.; Porri, F. Epigenetic Variation among Natural Populations of the South African Sandhopper Talorchestia Capensis. Evol. Ecol. 2017, 31, 77–91.
- Whitaker, J.M.; Welsh, A.B.; Hondorp, D.W.; Boase, J.C.; Merovich, G.T.; Welsh, S.; Krueger, C. Variation in DNA Methylation Is Associated with Migratory Phenotypes of Lake Sturgeon Acipenser fulvescens in the St. Clair River, MI, USA. J. Fish Biol. 2018, 93, 942–951.
- Gavery, M.R.; Nichols, K.M.; Goetz, G.W.; Middleton, M.A.; Swanson, P. Characterization of Genetic and Epigenetic Variation in Sperm and Red Blood Cells from Adult Hatchery and Natural-Origin Steelhead, Oncorhynchus Mykiss. G3 Genes|Genomes|Genet. 2018, 8, 3723–3736.
- Johnson, K.M.; Kelly, M.W. Population Epigenetic Divergence Exceeds Genetic Divergence in the Eastern Oyster Crassostrea virginica in the Northern Gulf of Mexico. Evol. Appl. 2020, 13, 945–959.
- Wogan, G.O.U.; Yuan, M.L.; Mahler, D.L.; Wang, I.J. Genome-wide Epigenetic Isolation by Environment in a Widespread Anolis Lizard. Mol. Ecol. 2020, 29, 40–55.
- Watson, H.; Powell, D.; Salmón, P.; Jacobs, A.; Isaksson, C. Urbanization Is Associated with Modifications in DNA Methylation in a Small Passerine Bird. Evol. Appl. 2021, 14, 85–98.
- McNew, S.M.; Beck, D.; Sadler-Riggleman, I.; Knutie, S.A.; Koop, J.A.H.; Clayton, D.H.; Skinner, M.K. Epigenetic Variation between Urban and Rural Populations of Darwin’s Finches. BMC Evol. Biol. 2017, 17, 183.
- Thorson, J.L.M.; Smithson, M.; Sadler-Riggleman, I.; Beck, D.; Dybdahl, M.; Skinner, M.K. Regional Epigenetic Variation in Asexual Snail Populations among Urban and Rural Lakes. Environ. Epigenetics 2019, 5, dvz020.
- Wang, X.; Li, A.; Wang, W.; Zhang, G.; Li, L. Direct and Heritable Effects of Natural Tidal Environments on DNA Methylation in Pacific Oysters (Crassostrea Gigas). Environ. Res. 2021, 197, 111058.
- Flatscher, R.; Frajman, B.; Schönswetter, P.; Paun, O. Environmental Heterogeneity and Phenotypic Divergence: Can Heritable Epigenetic Variation Aid Speciation? Genet. Res. Int. 2012, 2012, 1–9.
- Smith, T.A.; Martin, M.D.; Nguyen, M.; Mendelson, T.C. Epigenetic Divergence as a Potential First Step in Darter Speciation. Mol. Ecol. 2016, 25, 1883–1894.
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic Variation in Mangrove Plants Occurring in Contrasting Natural Environment. PLoS ONE 2010, 5, e10326.
- Medrano, M.; Herrera, C.M.; Bazaga, P. Epigenetic Variation Predicts Regional and Local Intraspecific Functional Diversity in a Perennial Herb. Mol. Ecol. 2014, 23, 4926–4938.
- Foust, C.M.; Preite, V.; Schrey, A.W.; Alvarez, M.; Robertson, M.H.; Verhoeven, K.J.F.; Richards, C.L. Genetic and Epigenetic Differences Associated with Environmental Gradients in Replicate Populations of Two Salt Marsh Perennials. Mol. Ecol. 2016, 25, 1639–1652.
- Morán, P.; Pérez-Figueroa, A. Methylation Changes Associated with Early Maturation Stages in the Atlantic Salmon. BMC Genet. 2011, 12, 86.
- Skinner, M.K.; Gurerrero-Bosagna, C.; Haque, M.M.; Nilsson, E.E.; Koop, J.A.H.; Knutie, S.A.; Clayton, D.H. Epigenetics and the Evolution of Darwin’s Finches. Genome Biol. Evol. 2014, 6, 1972–1989.
- Liu, S.; Sun, K.; Jiang, T.; Feng, J. Natural Epigenetic Variation in Bats and Its Role in Evolution. J. Exp. Biol. 2015, 218, 100–106.
- Zhang, X.; Li, Q.; Kong, L.; Yu, H. Epigenetic Variation of Wild Populations of the Pacific Oyster Crassostrea Gigas Determined by Methylation-Sensitive Amplified Polymorphism Analysis. Fish Sci. 2018, 84, 61–70.
- Vernaz, G.; Malinsky, M.; Svardal, H.; Du, M.; Tyers, A.M.; Santos, M.E.; Durbin, R.; Genner, M.J.; Turner, G.F.; Miska, E.A. Mapping Epigenetic Divergence in the Massive Radiation of Lake Malawi Cichlid Fishes. Nat. Commun. 2021, 12, 5870.
- Massicotte, R.; Whitelaw, E.; Angers, B. DNA Methylation: A Source of Random Variation in Natural Populations. Epigenetics 2011, 6, 421–427.
- Massicotte, R.; Angers, B. General-Purpose Genotype or How Epigenetics Extend the Flexibility of a Genotype. Genet. Res. Int. 2012, 2012, 317175.
- Thorson, J.L.M.; Smithson, M.; Beck, D.; Sadler-Riggleman, I.; Nilsson, E.; Dybdahl, M.; Skinner, M.K. Epigenetics and Adaptive Phenotypic Variation between Habitats in an Asexual Snail. Sci. Rep. 2017, 7, 14139.
- Berbel-Filho, W.M.; Rodríguez-Barreto, D.; Berry, N.; Garcia De Leaniz, C.; Consuegra, S. Contrasting DNA Methylation Responses of Inbred Fish Lines to Different Rearing Environments. Epigenetics 2019, 14, 939–948.
- Richards, E.J. Inherited Epigenetic Variation—Revisiting Soft Inheritance. Nat. Rev. Genet. 2006, 7, 395–401.
- Sheldon, E.L.; Schrey, A.; Andrew, S.C.; Ragsdale, A.; Griffith, S.C. Epigenetic and Genetic Variation among Three Separate Introductions of the House Sparrow (Passer domesticus) into Australia. R. Soc. Open Sci. 2018, 5, 172185.
- Fargeot, L.; Loot, G.; Prunier, J.G.; Rey, O.; Veyssière, C.; Blanchet, S. Patterns of Epigenetic Diversity in Two Sympatric Fish Species: Genetic vs. Environmental Determinants. Genes 2021, 12, 107.
- McRae, A.F.; Powell, J.E.; Henders, A.K.; Bowdler, L.; Hemani, G.; Shah, S.; Painter, J.N.; Martin, N.G.; Visscher, P.M.; Montgomery, G.W. Contribution of Genetic Variation to Transgenerational Inheritance of DNA Methylation. Genome Biol. 2014, 15, R73.
- Carja, O.; MacIsaac, J.L.; Mah, S.M.; Henn, B.M.; Kobor, M.S.; Feldman, M.W.; Fraser, H.B. Worldwide Patterns of Human Epigenetic Variation. Nat. Ecol. Evol. 2017, 1, 1577–1583.
- Orozco, L.D.; Morselli, M.; Rubbi, L.; Guo, W.; Go, J.; Shi, H.; Lopez, D.; Furlotte, N.A.; Bennett, B.J.; Farber, C.R.; et al. Epigenome-Wide Association of Liver Methylation Patterns and Complex Metabolic Traits in Mice. Cell Metab. 2015, 21, 905–917.
- Liebl, A.L.; Schrey, A.W.; Richards, C.L.; Martin, L.B. Patterns of DNA Methylation Throughout a Range Expansion of an Introduced Songbird. Integr. Comp. Biol. 2013, 53, 351–358.
- Leung, C.; Breton, S.; Angers, B. Facing Environmental Predictability with Different Sources of Epigenetic Variation. Ecol. Evol. 2016, 6, 5234–5245.
- Wenzel, M.A.; Piertney, S.B. Fine-Scale Population Epigenetic Structure in Relation to Gastrointestinal Parasite Load in Red Grouse (Lagopus lagopus scotica). Mol. Ecol. 2014, 23, 4256–4273.
- Venney, C.J.; Sutherland, B.J.G.; Beacham, T.D.; Heath, D.D. Population Differences in Chinook Salmon (Oncorhynchus tshawytscha) DNA Methylation: Genetic Drift and Environmental Factors. Ecol. Evol. 2021, 11, 6846–6861.
- Slotkin, R.K.; Martienssen, R. Transposable Elements and the Epigenetic Regulation of the Genome. Nat. Rev. Genet. 2007, 8, 272–285.
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as Regulators of Gene Expression. Science 2016, 351, aac7247.
- Schauer, S.N.; Carreira, P.E.; Shukla, R.; Gerhardt, D.J.; Gerdes, P.; Sanchez-Luque, F.J.; Nicoli, P.; Kindlova, M.; Ghisletti, S.; Santos, A.D.; et al. L1 Retrotransposition Is a Common Feature of Mammalian Hepatocarcinogenesis. Genome Res. 2018, 28, 639–653.
- Liu, J.; Ali, M.; Zhou, Q. Establishment and Evolution of Heterochromatin. Ann. N. Y. Acad. Sci. 2020, 1476, 59–77.
- Platt, A.; Gugger, P.F.; Pellegrini, M.; Sork, V.L. Genome-Wide Signature of Local Adaptation Linked to Variable CpG Methylation in Oak Populations. Mol. Ecol. 2015, 24, 3823–3830.
- Macia, A.; Muñoz-Lopez, M.; Cortes, J.L.; Hastings, R.K.; Morell, S.; Lucena-Aguilar, G.; Marchal, J.A.; Badge, R.M.; Garcia-Perez, J.L. Epigenetic Control of Retrotransposon Expression in Human Embryonic Stem Cells. Mol. Cell. Biol. 2011, 31, 300–316.
- Tang, M.-H.; Varadan, V.; Kamalakaran, S.; Zhang, M.Q.; Dimitrova, N.; Hicks, J. Major Chromosomal Breakpoint Intervals in Breast Cancer Co-Localize with Differentially Methylated Regions. Front. Oncol. 2012, 2, 197.
- Makova, K.D.; Hardison, R.C. The Effects of Chromatin Organization on Variation in Mutation Rates in the Genome. Nat. Rev. Genet. 2015, 16, 213–223.
- Duncan, B.K.; Miller, J.H. Mutagenic Deamination of Cytosine Residues in DNA. Nature 1980, 287, 560–561.
- Britten, R.J.; Baron, W.F.; Stout, D.B.; Davidson, E.H. Sources and Evolution of Human Alu Repeated Sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 4770–4774.
- Kilvitis, H.J.; Hanson, H.; Schrey, A.W.; Martin, L.B. Epigenetic Potential as a Mechanism of Phenotypic Plasticity in Vertebrate Range Expansions. Integr. Comp. Biol. 2017, 57, 385–395.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
760
Revisions:
2 times
(View History)
Update Date:
19 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No