 +1 credit
+1 credit
Video Upload Options
The ATPase family AAA-domain containing protein 3A (ATAD3A), a nuclear-encoded mitochondrial enzyme, contributes to mitochondrial dynamics, nucleoid organization, protein translation, cell growth, and cholesterol metabolism. The ATAD3A protein contains two coiled-coil domains (CC1 and CC2), Walker A (WA) and Walker B (WB) motifs and among them, the WA motif is responsible for ATP binding in the AAA module of ATAD3A. ATAD3A is an understudied protein in cancer, although we have demonstrated it functions as a metastasis promoter in breast cancer. At this stage of our understanding, ATAD3A dysfunction is also required and sufficient to drive oncogenic process in many types of cancer. Thus, there is a need to understand the mechanism by which ATAD3A interacts with other mitochondria-localized oncoproteins, and the targeting strategy in which ATAD3A is abrogated.
1. Introduction
In addition to their bioenergetic role, mitochondria function as signaling platforms and key regulators of cellular processes related to biosynthesis, Ca2+ homeostasis, and cell death. In cancer, mitochondrial function is critical to cell survival through genetic and/or environmental events, leading to metabolic reprogramming and changes in mitochondrial biogenesis, mitophagy, and dynamics. While the function of mitochondria in cancer has historically been restricted to Warburg’s hypothesis (aerobic glycolysis), work over recent decades has served to help dispel misconceptions and deepen our understanding of the diverse and dynamic roles that these organelles play throughout tumor progression, such as in cell survival, proliferation, stemness, motility, metastasis, and therapeutic resistance [1][2]. Non-specific targeting of mitochondrial functions in the treatment of cancer, however, may have major unwarranted effects, like inhibition of normal cell growth. Therefore, refined strategies that allow for the specific functional blocking of oncoproteins that physically localize to the mitochondria in cancer cells will have to be devised for therapeutic intervention. Yet, given that there is no simple canon for the role of mitochondrial oncoproteins in the regulation of malignant mitochondrial programs, gaining mechanistic insights into these proteins and their respective signaling networks involved in tumor development and progression will be critical to the clinical exploration of novel anticancer therapies.
Mitochondrial ATPase family AAA domain-containing protein 3 (ATAD3) belongs to AAA+ (ATPases associated with various cellular activities) superfamily, which shares a highly conserved module for ATP hydrolysis and participates in a variety of cellular processes [3][4]. ATAD3 only exists in eukaryotic organisms and has three family members: ATAD3A, ATAD3B, and ATAD3C. Among them, ATAD3B and ATAD3C only exist in primates and humans. While, ATAD3B may act as a dominant negative inhibitor to ATAD3A function [5], the exact role of ATAD3C remains unknown. ATAD3A is believed to be the ancestral form and to be duplicated twice to form ATAD3B and ATAD3C [6]. Structurally, the ATAD3A protein spans the mitochondrial outer membrane (OM) and inner membrane (IM) and regulates dynamic interactions between the two that is sensed by cell fission machinery [4]. As a mitochondrial protein with the capacity to impact essential mitochondrial functions and organization, ATAD3A controls a broad spectrum of physiological and pathological responses, including mitochondrial dynamics, nucleoid organization, signaling transduction, and cholesterol metabolism [7][8][9]. ATAD3A mutations can cause a range of different phenotypes and have been identified as one of the most common causes of lethal infantile mitochondrial disease [10]. Although there have been no or few ATAD3A mutations identified in cancer patients, ATAD3A has nevertheless been implicated in certain types of cancer, where its elevated expression levels have been associated with poor patient outcome.
2. The Essential Role of ATAD3A in Mitochondria
2.1. The Molecular Structure of ATAD3A in Mitochondria
ATAD3A is located on chromosome 1 at 1p36.33 locus and has three transcript variants, with isoform 2 being the major one with 586 amino acids (a.a.) (Figure.1). While the first 50 amino acids of the protein’s N-terminal can be found on the mitochondrial surface, there are several important domains within the N-terminal, including a flexible proline-rich region for possible protein–protein interactions (a.a. 18–27), transmembrane domain 1 (TM1, a.a. 225–242) for integrating the mitochondrial OM, transmembrane domain 2 (TM2, a.a. 247–264) for integrating the mitochondrial IM, and two coiled-coil regions (CC1, a.a. 85 to 115; CC2, a.a. 180–220) for the oligomerization of ATAD3A monomers and/or for interaction with other proteins [11]. Deacetylation on lysine 135 (K135) residue of ATAD3A is required for its oligomerization, especially for dimerization [12]. The ATPase domain and two ATP binding domains, Walker A (WA) and Walker B (WB), are located at the C-terminal of ATAD3A [5][13] (Figure 1). In particular, mutations on K355 or K358 of the WA domain can block the binding of ATPs to ATAD3A, subsequently influencing ATAD3A’s ATP affinity and reducing its ATPase activity [14]. Of note, these two mutations have been found to be disease-relevant and dominantly inherited in a family with hereditary spastic paraplegia [14]. However, high levels of ATAD3A expression, rather than ATAD3A mutations, have historically been identified in cancer patients [4]. Aside, it is worth mentioning that both the N-terminal and C-terminal regions of ATAD3A have been suggested to contribute to the protein’s interaction with S100B, a zinc and calcium-binding protein with a chaperone-associated function contributing to proper ATAD3A protein folding [15]. Interestingly, while the role of S100B in cancer is not yet well understood, interrogation of its interactions with target proteins like p53 [16] and the potential to serve as a marker for metastasis in different cancers has been reported [17].
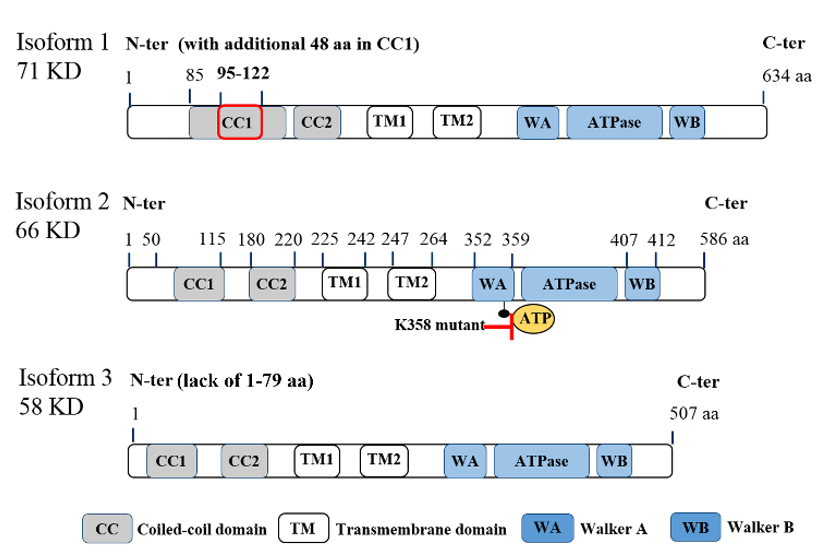
Figure 1. Molecular structure of ATPase family AAA domain-containing protein 3A (ATAD3A) transcript variants with vital domains. Schematic diagram for the molecular structure of three transcript variants of ATAD3A. Three isoforms have similar major domains, including two coiled-coil domains (CC1 and CC2), two transmembrane domains (TM1 and TM2), walker A (WA) and walker B (WB) domains for the ATP binding, and ATPase domain. Isoform 2 (66KD) is the major form in cancer cells. Compared with isoform 2, isoform 1 has an additional 48 amino acids in the CC1 domain, which may disrupt its function to form the oligomers or interaction with other mitochondrial partners. Isoform 3 lacks the first 50 amino acids in the N-terminal, which locates on the mitochondria surface and is essential for the interaction with cytoplasmic proteins. Of note, mutations on K355 or K358 in the WA domain markedly reduce the ATPase activity of ATAD3A.
2.2. Functions of ATAD3A in Mitochondria Homeostasis
Mitochondria function is tightly associated with its dynamics, including mitochondrial fission and fusion. ATAD3A regulates mitochondrial dynamics through its interactions with mitochondrial fission (dynamin-related protein 1, DRP1) and fusion (mitofusins, OPA1) proteins [18] (Figure 2). Silencing ATAD3A by small interfering RNAs (siRNAs) or ectopic expression of deficient mutant ATAD3A increases mitochondrial fragmentation, while knockdown of DRP1 eliminates mitochondrial fragmentation [14][19]. In addition to its involvement with mitochondrial fusion and shaping, mitofusin-2 is critical for maintaining close mitochondrial interaction with the endoplasmic reticulum (ER) [20]; when mitofusion-2 is depleted, ATAD3A localization to mitochondrial-associated membranes is increased [21]. As an ATP-dependent chaperonin, HSP60 mediates mitochondrial proteostasis with its co-chaperonin HSP10. In particular, the interaction of ATAD3A and HSP60 has been detected at ATAD3A’s C-terminal [8] (Figure 2). ATP binding-deficient ATAD3A harbors a mutation in its WA domain, leading to mitochondrial fragmentation in glioblastoma cells [8].
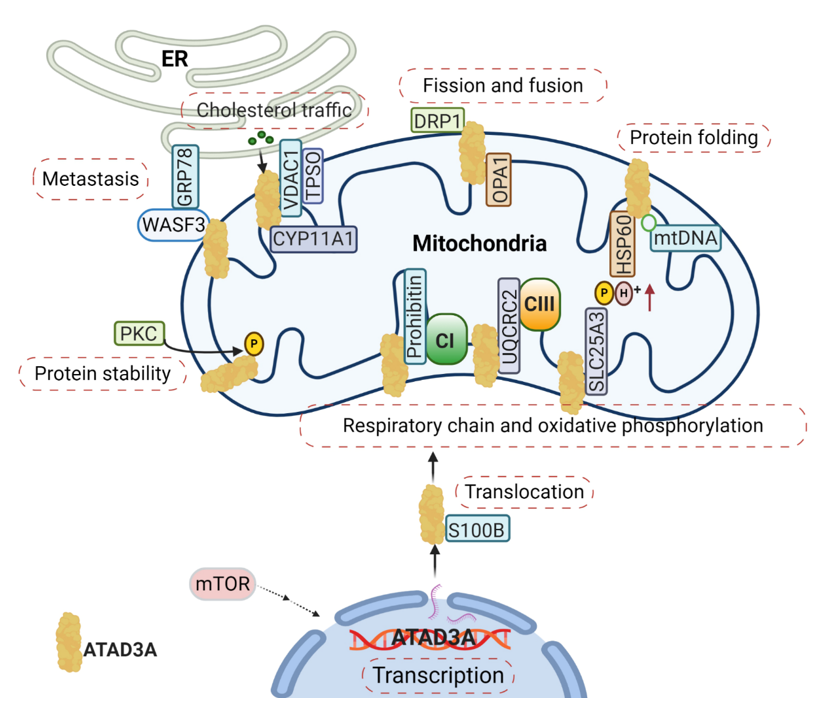
Figure 2. The complexity of the ATAD3A signaling network in mitochondria. The main signaling pathways involve (1) the regulation of ATAD3A expression levels by mTOR, (2) the proper folding and mitochondrial localization of newly synthesized ATAD3A protein associated with S100B’s function, (3) ATAD3A protein stability regulation by PKC, (4) and communication between the endoplasmic reticulum (ER) and mitochondria mediated by the ATAD3A/WASF3/GPR78 axis. Moreover, ATAD3A has been identified as one essential part of transduceosome, also known as the cholesterol transfer complex. ATAD3A has the ability to assist with the transportation and metabolism of cholesterol by interacting with voltage-dependent anion channel (VDAC) and CYP11A1 in mitochondria. ATAD3A governs mitochondrial dynamics through the functional regulations of mitochondrial fission- dynamin-related protein 1 (DRP1) and fusion protein-OPA1. ATAD3A also contributes to mitochondrial respiration via interactions with several important respiration proteins, such as prohibitin, UQCRC2 and SLC25A3.
Mitochondrial DNA (mtDNA) is organized in the nucleoprotein complex associated with the IMM. mtDNA segregation and changes in mitochondrial architecture can be induced by altering the structure or composition of the nucleoid [22]. Interestingly, altered ATAD3A expression perturbs mtDNA maintenance and replication [23]. A more recent study shows that ATAD3A is a detergent-resistant component that organizes mtDNA and segregates mitochondrial nucleoids, and that ATAD3A deficiency leads to modifications in mtDNA organization [24]. Moreover, loss of ATAD3A induces early and severe mitochondrial structural abnormalities, progressive mtDNA depletion and deletions, and muscle atrophy in mice [25]. Loss of ATAD3A also leads to a dramatic reduction in mitochondrial cristae junctions and changes in cristae morphology [25]. Further, ATAD3A plays a critical role in regulating IMM structure, leading to secondary defects in mtDNA replication, complex V, and cholesterol levels [25]. Interestingly, in later work evaluating human siblings with a recessive missense ATAD3A mutation that likely disrupts WB, it was found that while the patient’s fibroblast possessed mitochondrial cristae malformations alongside decreased ATAD3A levels, in contrast to knockout mouse models, no changes in oxidative phosphorylation complexes were seen [26]. Importantly, ATAD3A has been reported to maintain homeostasis in mouse hematopoietic cells by impeding Pink1 mitophagy. Here, it was seen that the deletion of ATAD3A hyperactivates mitophagy by facilitating Pink1 transportation and activity [27].
Mitochondria–ER interactions are critical to enabling mitochondrial adaptation and maintaining organelle homeostasis. Particularly, the ER supplies biomolecules needed for biogenesis and helps govern numerous processes, such as those involved in stress and morphological changes [28]. Given the critical link between these two organelles, it has been suggested that ATAD3, as a contact site, could represent an evolutionary step towards mitochondrial adaptation to ER interactions [29]. Nevertheless, ATAD3A’s various involvements and precise functions have yet to be fully elucidated.
2.3. The Role of ATAD3A in Mitochondrial Metabolism and Respiration
From Nematoda to mammalian, ATAD3A is critical in the development of a number of multicellular organisms. Silencing ATAD3A in C. elegans and Drosophila induces growth arrest in larvae [7][8]. In murine embryos, knockout of ATAD3A is lethal, causing retardation and defects in trophoblast lineages, possibly due to low mitochondrial biogenesis and ATP production [9]. Deletion or mutation of ATAD3A in the WA domain has also been linked to distinct neurological syndromes in humans, including global developmental delay, hypotonia, optic atrophy, axonal neuropathy, and hypertrophic cardiomyopathy [14][30]. Steroid hormones are synthesized in the mitochondria and smooth ER by a variety of tissues, such as the adrenal cortex, gonads, and placenta. These hormones are all derived from cholesterol and influence the development and progression of human cancers by binding steroid hormone receptors (SHRs) [31][32]. Cholesterol trafficking occurs between the ER and mitochondria, where communication between the two organelles facilitates both steroidogenesis substrate availability and mitochondria product passage to different steroidogenic enzymes in the ER [33]. During steroidogenesis, the rate-limiting step is the transfer of cholesterol from the OMM to the IMM, where it is converted into pregnenolone by the cytochrome P450 enzyme CYP11A1 [34] (Figure 2). Alongside the voltage-dependent anion channel (VDAC) and other constituents like cytosolic proteins, ATAD3A has been identified as an essential component of the transduceosome complex through which this transport of cholesterol is facilitated [34] (Figure 2).
In MA-10 mouse tumor Leydig cells, knockdown of ATAD3A leads to a significant decrease in steroid production [33]; and in patients with ATAD3 gene cluster deletions, derived fibroblasts display abnormalities in cholesterol metabolism [24]. Our studies show that the ATAD3A-WASF3-GRP78 axis, which bridges the interaction between the mitochondria and ER, may possess a potential role in the regulation of cholesterol traffic [35]. In addition, the critical role of ATAD3A in mitochondria metabolism, especially of lipids, has been confirmed in several kinds of model organisms. In C. elegans, reduction of intestinal fat storage and low lysosomal content have been reported when ATAD3A is knocked down [7]. Interestingly, it has been revealed that both the N-terminal and C-terminal of ATAD3A are required for normal cell growth and cholesterol channeling in Drosophila [8]. Lastly, altered cholesterol metabolism was reported in the skeletal muscle of conditional ATAD3A knockout mice [25].
It has also been demonstrated that ATAD3A participates in mitochondrial respiration [27]. In C. elegans, silencing ATAD3A decreases levels of both complex I and citrate synthase, diminishing mitochondrial activity and ultimately impeding larval development [7]. In mouse hematopoietic cells, knockout of ATAD3A results in decreased mitochondrial mass and impaired mitochondrial functions, with abnormalities seen through lower rates of basal oxygen-consumption and diminished oxidative capacity [27]. Several interactions between ATAD3A and components critical to mitochondrial respiration have been identified and include prohibitin, UQCRC2, and SLC25A3 [21] (Figure 2). Prohibitin associates and stabilizes respiratory complexes, particularly Complex I, and regulates the proteolysis of unassembled IMM proteins of the oxidative phosphorylation system [36]. As a core subunit of Complex III, UQCRC2 is needed for complex III’s conversion into its catalytically active homodimer form, which can subsequently be incorporated into a larger supercomplex that functions as one enzyme [37]. SLC25A3 is located in the IMM and serves to transports phosphate groups (along with H+) from the cytosol to the mitochondrial matrix during oxidative phosphorylation [38]. The ways by which ATAD3A regulates mitochondrial respiration through these protein interactions, however, remains unclear. Notably, numerous other respiratory complex components have been found to be directly or indirectly related to ATAD3A, further obscuring the protein’s exact roles. Particularly, ATAD3A knockdown in mouse 3T3-L is seen to decrease the expression, for example, of MTCO1, MTCO2, ATP5A, UQCRC2, SDHB, NDUFB8, and NDUFA10 [33].
References
- Porporato, E.; Filigheddu, N.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280.
- Grasso, ; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146.
- Wendler, ; Ciniawsky, S.; Kock, M.; Kube, S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 2–14.
- Teng, ; Lang, L.; Shay, C. ATAD3A on the Path to Cancer. In Reviews on Biomarker Studies of Metabolic and Metabolism-Related Disorders; Springer: Berlin/Heidelberg, Germany, 2019; pp. 259–269.
- Merle, ; Féraud, O.; Gilquin, B.; Hubstenberger, A.; Kieffer-Jacquinot, S.; Assard, N.; Bennaceur-Griscelli, A.; Honnorat, J.; Baudier, J. ATAD3B is a human embryonic stem cell specific mitochondrial protein, re-expressed in cancer cells, that functions as dominant negative for the ubiquitous ATAD3A. Mitochondrion 2012, 12, 441–448.
- Li, ; Rousseau, D. ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J. Bioenerg. Biomembr. 2012, 44, 189–197.
- Hoffmann, ; Bellance, N.; Rossignol, R.; Koopman, W.J.; Willems, P.H.; Mayatepek, E.; Bossinger, O.; Distelmaier, F. C. elegans ATAD-3 is essential for mitochondrial activity and development. PLoS ONE 2009, 4, e7644.
- Gilquin, ; Taillebourg, E.; Cherradi, N.; Hubstenberger, A.; Gay, O.; Merle, N.; Assard, N.; Fauvarque, M.-O.; Tomohiro, S.; Kuge, O. The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol. Cell. Biol. 2010, 30, 1984–1996.
- Goller, ; Seibold, U.K.; Kremmer, E.; Voos, W.; Kolanus, W. Atad3 function is essential for early post-implantation development in the mouse. PLoS ONE 2013, 8, e54799.
- Frazier, E.; Compton, A.G.; Kishita, Y.; Hock, D.H.; Welch, A.E.; Amarasekera, S.S.; Rius, R.; Formosa, L.E.; Imai-Okazaki, A.; Francis, D. Fatal Perinatal Mitochondrial Cardiac Failure Caused by Recurrent De Novo Duplications in the ATAD3 Locus. Med 2020, 1, 1–25.
- Baudier, ATAD3 proteins: Brokers of a mitochondria–endoplasmic reticulum connection in mammalian cells. Biol. Rev. 2017, 93, 827–844.
- Zhao, ; Sun, X.; Hu, D.; Prosdocimo, D.A.; Hoppel, C.; Jain, M.K.; Ramachandran, R.; Qi, X. ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat. Commun. 2019, 10, 1371.
- Hubstenberger, ; Merle, N.; Charton, R.; Brandolin, G.; Rousseau, D. Topological analysis of ATAD3A insertion in purified human mitochondria. J. Bioenerg. Biomembr. 2010, 42, 143–150.
- Cooper, M.; Yang, Y.; Ylikallio, E.; Khairullin, R.; Woldegebriel, R.; Lin, K.-L.; Euro, L.; Palin, E.; Wolf, A.; Trokovic, R. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 2017, 26, 1432–1443.
- Gilquin, ; Cannon, B.R.; Hubstenberger, A.; Moulouel, B.; Falk, E.; Merle, N.; Assard, N.; Kieffer, S.; Rousseau, D.; Wilder, P.T. The calcium-dependent interaction between S100B and the mitochondrial AAA ATPase ATAD3A and the role of this complex in the cytoplasmic processing of ATAD3A. Mol. Cell. Biol. 2010, 30, 2724–2736.
- Baudier, ; Deloulme, J.C.; Shaw, G.S. The Zn2+ and Ca2+-binding S100B and S100A1 proteins: Beyond the myths. Biol. Rev. 2020, 95, 738–758.
- Yen, -C.; Huang, Y.-C.; Kan, J.-Y.; Kuo, P.-L.; Hou, M.-F.; Hsu, Y.-L. S100B expression in breast cancer as a predictive marker for cancer metastasis. Int. J. Oncol. 2018, 52, 433–440.
- Fang, -Y.; Chang, C.-L.; Hsu, S.-H.; Huang, C.-Y.; Chiang, S.-F.; Chiou, S.-H.; Huang, C.-H.; Hsiao, Y.-T.; Lin, T.-Y.; Chiang, I.-P. ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci. 2010, 123, 1171–1180.
- Spelbrink, N. Functional organization of mammalian mitochondrial DNA in nucleoids: History, recent developments, and future challenges. IUBMB Life 2010, 62, 19–32.
- Naon, ; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254.
- Chiang, -F.; Huang, C.-Y.; Lin, T.-Y.; Chiou, S.-H.; Chow, K.-C. An alternative import pathway of AIF to the mitochondria. Int. J. Mol. Med. 2012, 29, 365–372.
- Holt, J.; He, J.; Mao, C.-C.; Boyd-Kirkup, J.D.; Martinsson, P.; Sembongi, H.; Reyes, A.; Spelbrink, J.N. Mammalian mitochondrial nucleoids: Organizing an independently minded genome. Mitochondrion 2007, 7, 311–321.
- He, ; Cooper, H.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.; Gao, J.; Neuman, K.; Fearnley, I.M.; Spinazzola, A. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121.
- Desai, ; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610.
- Peralta, ; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismäki, J.; Moraes, C.T. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 2018, 131, jcs217075.
- Peralta, ; González-Quintana, A.; Ybarra, M.; Delmiro, A.; Pérez-Pérez, R.; Docampo, J.; Arenas, J.; Blázquez, A.; Ugalde, C.; Martín, M.A. Novel ATAD3A recessive mutation associated to fatal cerebellar hypoplasia with multiorgan involvement and mitochondrial structural abnormalities. Mol. Genet. Metab. 2019, 128, 452–462.
- Jin, ; Xu, C.; Zhang, X.; Long, J.; Rezaeian, A.H.; Liu, C.; Furth, M.E.; Kridel, S.; Pasche, B.; Bian, X.-W. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 2018, 19, 29.
- Szymański, ; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 2017, 18, 1576.
- Rousseau, ATAD3 and endoplasmic reticulum to mitochondria connection: A main actor and interaction regarding pathogenesis. J. Med. 2019, 3, 1–7.
- Harel, ; Yoon, W.H.; Garone, C.; Gu, S.; Coban-Akdemir, Z.; Eldomery, M.K.; Posey, J.E.; Jhangiani, S.N.; Rosenfeld, J.A.; Cho, M.T. Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am. J. Hum. Genet. 2016, 99, 831–845.
- Ahmad, ; Kumar, R. Steroid hormone receptors in cancer development: A target for cancer therapeutics. Cancer Lett. 2011, 300, 1–9.
- Holst, P.; Soldin, O.P.; Guo, T.; Soldin, S.J. Steroid hormones: Relevance and measurement in the clinical laboratory. Clin. Lab. Med. 2004, 24, 105–118.
- Issop, ; Fan, J.; Lee, S.; Rone, M.B.; Basu, K.; Mui, J.; Papadopoulos, V. Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: Role of ATAD3. Endocrinology 2014, 156, 334–345.
- Rone, B.; Midzak, A.S.; Issop, L.; Rammouz, G.; Jagannathan, S.; Fan, J.; Ye, X.; Blonder, J.; Veenstra, T.; Papadopoulos, V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol. Endocrinol. 2012, 26, 1868–1882.
- Teng, ; Pi, W.; Wang, Y.; Cowell, J.K. WASF3 provides the conduit to facilitate invasion and metastasis in breast cancer cells through HER2/HER3 signaling. Oncogene 2016, 35, 4633–4640.
- Signorile, ; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A critical role in mitochondrial functions and implication in diseases. Cells 2019, 8, 71.
- Miyake, ; Yano, S.; Sakai, C.; Hatakeyama, H.; Matsushima, Y.; Shiina, M.; Watanabe, Y.; Bartley, J.; Abdenur, J.E.; Wang, R.Y. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum. Mutat. 2013, 34, 446–452.
- Boulet, ; Vest, K.E.; Maynard, M.K.; Gammon, M.G.; Russell, A.C.; Mathews, A.T.; Cole, S.E.; Zhu, X.; Phillips, C.B.; Kwong, J.Q. The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem. 2018, 293, 1887–1896.


