 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio E Pontiroli | + 4829 word(s) | 4829 | 2020-11-03 08:49:00 | | | |
| 2 | Bruce Ren | -1746 word(s) | 3083 | 2020-11-12 02:41:55 | | | | |
| 3 | Bruce Ren | + 3 word(s) | 3086 | 2020-11-12 03:07:17 | | | | |
| 4 | Bruce Ren | Meta information modification | 3086 | 2020-11-23 08:43:45 | | | | |
| 5 | Lucia La Sala | Meta information modification | 3086 | 2020-11-24 22:14:20 | | |
Video Upload Options
Obesity is one of the major risk factors for the development of both impaired glucose tolerance (IGT, or prediabetes) and type 2 diabetes (T2D), and its prevalence worldwide drives toward an increased rate of cardiovascular morbidity and mortality. Given the estimations of the World Health Organization (WHO) and the recommendation of the Diabetes Prevention Program (DPP), where IGT and diabetes are considered as risk factors for the development of cardiovascular complications and obesity, the development of diabetes should be treated because of its potential reversibility. In this view, several interventions such as diet, lifestyle changes, and pharmacological treatment are effective, including bariatric metabolic surgery (BMS), which is the most incisive way to efficiently lower body weight. Here the attention is focused on links between obesity, T2D, and cardiovascular disease, as possible hints to possible new treatments.
1. Introduction
The prevalence of obesity has risen dramatically worldwide over the last decades, reaching epidemic proportions. It has been estimated by the World Health Organization (WHO) that the number of obese adults has increased more than 7-fold in the last 40 years [1]. Obesity is associated with a high prevalence of impaired glucose tolerance (IGT, or prediabetes), and it is an independent risk factor for type 2 diabetes (T2D) [2], as shown by data in the general population and in patients who are candidates for bariatric surgery [3][4]; in turn, prediabetes is associated with the risk of progression to diabetes, and it has been recognized for a long time that prediabetes unfavorably affects the cardiovascular system [5]. Growing evidence has shown a dramatic increase in overweight/obesity prevalence also among adolescents. Recently, a cohort study on an adolescent population has evidenced that increased body mass index (BMI) is associated with a higher risk to develop T2D in the adult life [6]. Other data regarding the reversibility of obesity in adolescents or young adults that in childhood were obese have focused on the reduction of the risk of developing T2D in adulthood [7]. Given these premises, it appears of utmost importance in obesity to prevent progression to diabetes, and in the meanwhile to treat co-morbidities of obesity, which are mainly represented by T2D, metabolic syndrome (MetS, a group of disorders strictly linked with each other and associated with insulin resistance (IR), elevated levels of triglycerides, and finally hyperglycemia), and cardiovascular disease (CVD). The Diabetes Prevention Program (DPP) recommended prevention programs as hints to reduce the risk to develop T2D, including adopting healthy habits (diet and physical activity) and avoiding smoking, alcohol, and stress. In fact, intensive lifestyle intervention was able to reduce the incidence of T2D by 58% over 3 years [8][9]. Therefore, preventing T2D in obesity seems to be critical for preventing metabolic diseases and cardiovascular complications.
2. Pathogenesis of Diabetes in Obesity
The pathogenesis of diabetes and obesity is similar, sharing common pathways of IR, oxidative stress (Ox-S), and pro-thrombotic and pro-inflammatory patterns [10][11]. The obesogenic environment, stimulating overnutrition, leads to dysregulation of the metabolic balance and subsequent fat accumulation in organs that are not specialized in lipid storage (ectopic fat), such as the endothelium, the liver, and the skeletal muscle, inducing metabolic disturbances and disorders, such as IR, IGT, and T2D, cardiovascular diseases, cerebrovascular diseases, and liver diseases. (Figure 1). Several mechanisms are strictly linked with the onset of CVD and atherosclerosis, in which endothelial dysfunction plays a major role [12].
2.1. Non-Esterified Fatty Acids (NEFAs) and Nitric Oxide (NO)
From an endothelial perspective, in obese people, the low levels of nitric oxide (NO) bioavailability lead to an impairment of endothelium-dependent vasodilation [13][14], with the consequent alteration of endothelial nitric oxide synthase (eNOS) and enhancement of Ox-S, inflammation, and atherosclerotic burden [11]. The mechanisms through which obesity leads to IR and subsequently to T2D are not fully understood. Several studies have shown in obesity that adipose tissue releases high amounts of circulating non-esterified fatty acids (NEFAs), hormones, and pro-inflammatory cytokines, which drive to IR by the inhibition of glucose transport and phosphorylation [15]. The increase in NEFAs provides the basis for the activation of the serine/threonine kinase cascade (induced by fatty acid metabolites such as ceramides, diacylglycerol, or fatty-acyl coenzyme A), leading to the phosphorylation of insulin receptor substrates 1 and 2 (IRS-1 and IRS-2). The ability of these receptor substrates can activate the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/eNOS pathway, reducing insulin receptor signaling and hence glucose transport. A hyperglycemic milieu could be worse from an endothelial perspective, especially in compromising the binding with the insulin receptor, activating a transduction way in favor of pro-atherogenic effects instead of anti-atherogenic responses. De Nigris et al. showed that under high levels of glucose and insulin-stimulated endothelial cells, the pro-atherogenic pathway seems to be more prone to activation [16]. In fact, the phosphorylation of eNOS at Ser1177, activated by AKT kinase, leads to the production of NO; but it also activates the phosphorylation of Src-homology 2 domain-containing (SHC) transforming protein, which affects the activation of the mitogen-activated protein kinase (MAPK) pathway, resulting in increased endothelin-1 (ET-1) expression and mitogenic effects [17].
2.2. Clock Genes
A piece of further evidence underlying the onset of T2D in obesity is represented by the alteration of the so-called "clock genes", which are considered as the precursors in the development of other metabolic complications. Circadian clocks are a group of genes responsible for coding the proteins needed for the generation and regulation of circadian rhythms, as demonstrated in both rodents [18] and humans [19]. The shift in circadian clock genes increases the risk of metabolic disorders, particularly obesity and IR. Several studies investigated the influence of the disturbance of clock gene expression on the development of obesity and diabetes. Various parameters related to glucose metabolism, such as glucose tolerance, insulin sensitivity, as well as glucose, glucagon, and insulin plasma levels are known to exhibit circadian variations throughout the day [20]. A dysregulation in the Circadian Locomotor Output Cycles Kaput (CLOCK) gene is able to disrupt the rhythm in feeding, promoting inactivity, enhancing hyperphagia, hyperlipidemia, hyperglycemia, and hypoinsulinemia [21]. Further studies focused on the Brain and Muscle Arnt-Like protein-1 (BMAL1) gene, which is a critical circadian transcription factor that encodes for protein Arnt-Like (ARNTL) forming a heterodimer with CLOCK. Mice lacking BMAL1 showed an alteration of adipogenesis and hepatic carbohydrate metabolism [22]. Otherwise, mice with inactivated BMAL1 showed a suppressed diurnal variation in glucose and triglycerides, which led to impaired gluconeogenesis and recovery from insulin-induced hypoglycemia [23]. In insulin-resistant mice, the perturbation of CLOCK, BMAL1, reduced c-erb-αoncogene (REV-ERBα) and Cryptochrome Circadian Clock 1 (CRY1) mRNA expression is evident during obesity [24]. Indeed, it seems that an activation of BMAL1, PPARα-induced, might have a direct link with high concentrations of retinoic acid (increased in metabolic syndrome [25]) and reduced REV-ERBα levels in obesity and IR [24]. The role of the transcription factor REV-ERBα gene, associated with MetS, was demonstrated by Vieira et al. in a study investigating clock gene expression in visceral adipose tissues (VATs) in lean and obese female subjects [26]. Furthermore, elucidating the role of clock genes might be helpful in the design of therapeutic interventions for obesity and T2D. For instance, the use of REV-ERB agonists in diet-induced obese mice increased energy expenditure, reduced fat mass, and improved dyslipidemia and hyperglycemia [27]. Furthermore, it has been proved that REV-ERB agonists would be beneficial in prevention or treatment models for atherosclerosis of LDL receptor null mice [28]. Due to its chemistry, REV-ERB ligands would be a target for the treatment of metabolic disease; thus, since high levels of REV-ERB have been correlated with increased insulin secretion and reduced glucose concentration, the use of REV-ERB agonists would be a potential treatment for T2D.
2.3. Insulin Resistance (IR) and Reactive Oxygen Species (ROS)
Obesity and T2D are closely associated with insulin resistance (IR) and the development of cardiovascular disorders (micro- and macrovascular), whose central factors are conveyed into processes of Ox-S [11]. Some studies have shown the role of Ox-S in the hyperglycemic phenotype and in a model of glucose variability using endothelial cells [29][30][31][32]. The glucose-derived cellular damage has been related to a hyperglycemia-induced overproduction of mitochondrial reactive oxygen species (ROS), resulting in defective ROS homeostasis and in the inactivation of antioxidant responses, in particular superoxide dismutase-2 (SOD-2) and glutathione peroxidase-1 (GPx-1), which are responsible for controlling the rate of radicals produced under stress conditions.
Obesity is well known to be affected by Ox-S having a higher susceptibility to activate oxidative pathways dysregulated by concurrent defective ROS scavenging and decreased mitochondrial function. The redox-sensitive transcription factor Nuclear Erythroid 2 related factor-2 (NFE2L2 or NRF2) regulates antioxidant response elements (AREs). In a diabetic milieu, the activity of NRF2 is reduced, contributing to increased Ox-S, mitochondrial dysfunction in vessels, and thus enhancing endothelial dysfunction, as observed in diabetes [33]. NRF2 activation is associated with the prevention of many types of human diseases, including diabetes and obesity [34]. In diabetic complications, elevated levels of circulating markers of lipid peroxidation were observed, reflecting the oxidative damage in several tissues [35].
2.4. Insulin Resistance (IR) and Inflammation
Both obesity and diabetes, in their pathogenesis and in the development of complications, share a common phenotype characterized by IR and the activation of inflammatory processes [36][37]. Many key inflammatory markers have been associated with both obesity and the risk of adverse outcomes in obesity-associated diseases, and this suggests that a persistent, low-grade inflammatory response is a potentially modifiable risk factor. The relationship between adipose tissue inflammation and glycemic control is complex. In fact, dysfunctional adiposity is characterized by an altered gene expression profile in the context of obesity and type 2 diabetes, which is not easy to identify [38].
An inflammatory role has been hypothesized in the pathways that drive toward the progression from obesity to diabetes. As demonstrated by an ARIC (Atherosclerosis Risk in Communities) study, a case-cohort study performed on diabetic and non-diabetic middle-aged subjects, inflammatory factors (a panel of six markers) have a major role in the pathways that lead to the progression from obesity to diabetes; in this population, obese individuals had a more than 6-fold higher risk of developing diabetes [39]. In ARIC, NEFAs was an independent predictor of diabetes and thus might play a mediating role; the hypothesized mechanisms focused on the involvement of an impaired insulin signaling pathway. In obese rats, the peripheral uptake of glucose in response to insulin was induced by the reduction of a pro-inflammatory cytokine, tumor necrosis factor-alpha (TNF-α) activity [40]. The underlying mechanism of the induction of IR by TNF-α has two different pathways:
- the activation of transcription factor NF-κB, which is involved in insulin-sensitivity, and Inhibitor of Nuclear Factor Kappa B Kinase Subunit β (IKKβ), and
- by directly inhibiting IRS-1 with phosphorylation on its serine residues [41].
Thus, the activated NF-κB promotes the transcription of multiple inflammatory mediators, such as TNF-α and interleukin-6 (IL-6) [42], promoting inflammation-induced IR pathway.
Furthermore, in non-obese diabetic (NOD) mice, autoimmunity has a prominent role, since inflammation can trigger the recruitment and activation of dendritic cells/macrophages, leading to the disruption of β-cells [43]. Finally, a possible role of resistin might be considered as a new link between obesity and diabetes, since resistin levels are increased in diet-induced obesity, as well as in genetic models of obesity and IR, and its inhibition enhanced insulin-stimulated glucose uptake [44]. Researchers also demonstrated the ability of the NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome, in which mRNA expression in VAT was correlated with body weight, to alter insulin sensitivity and IR through the processing of IL-1β and IL-18 in response to obesogenic stimuli as LDL, hypoxia, and ROS, promoting a chronic pro-inflammatory state [45].
3. Progression of IGT to T2D in obesity: from epigenetics to the role of miRNAs
The prevalence of diagnosed type 2 diabetes mellitus continued to increase concurrently with increases in obesity. Several experimental studies have highlighted the role of epigenetics in the control of progression towards pre-/diabetes. Mechanisms such as methylation of genes and expression of non-coding small RNA molecules (microRNAs) play a crucial role in these processes. microRNAs (miRs), a single-stranded small molecule of non-coding RNA highly conserved among species and regulating gene expression (as summarized in [46]), might be related to the progression of prediabetes towards diabetes: in DIAPASON study (detailed in [47]) it was possible to identify IGT progressors vs non-progressors, predicting diabetic progression in the pre-diabetic population [48]. Parrizas et al. proposed both miR-192 (which modulates adipocyte differentiation and lipid homeostasis in obese subjects [49]) and miR-193b (which modulates adiponectin production in white adipose tissue (WAT) and therapeutic target for IR [50]) as markers of prediabetes. Indeed, the downregulation of miR-192 only in diabetics suggested a possible therapeutic role to prevent diabetes [51]. As aforementioned, miRs are non-coding RNA molecules of 25 nucleotides in length able to modulate gene expression, and are emerging as new biomarkers for predicting diseases. A set of 24 circulating miRs selected by nutritional therapy based on low fat-high complex carbohydrate diet (LFHCC) recommended by American Heart Association (AHA), have emerged as preventive for T2D development in subjects with CVD [52]. Also, miR-21 in T2D subjects might be used as novel biomarker induced by elevated ROS that exert their dangerous activity on lipid peroxidation [31].
3.1. Novel miRNA-based approaches for the prevention of diabetes using bariatric metabolic surgery (BMS)
Bariatric metabolic surgery (BMS) has emerged as potentially useful for morbid obesity. BMS leads to remission/resolution of T2D, improving glycemic control [53] insulin sensitivity, hyperlipidemia and other obesity-associated disorders, but precise mechanisms are not yet fully understood. Recently, it has been demonstrated that the beneficial effects of BMS might be regulated by microRNAs that, due to their ability to bind the 3’-UTR of the mRNA target, modulate gene expression [54]. miRNAs have been related with the molecular changes typical of excessive adiposity, that could be resolved by weight loss after BMS. Analyzing miRNome from obese and normal-weight individuals some obesity-related changes in adipose tissue miRNome were observed: five miRs (hsa-miR-146b-3p, hsa-miR-146b-5p, hsa-miR-223-3p, hsa-miR-223-5p and hsa-miR-941) which were increased in obese subcutaneous adipose tissues (SAT-O), were significantly reduced after BMS [55].
Recently, in plasma and liver samples of both bariatric patients and a rodent model of BMS, it has been shown that a 90% downregulation of miR-122 and a reduction of miR-342-3p, of miR-320, of miR-139-5p and of miR-146a, might regulate metabolic processes as TCA cycle, glucose transport, pentose phosphate pathway, fatty-acid synthesis, mitochondrial oxidation, gluconeogenesis and glycolysis [56][57]. Besides, it has been demonstrated that BMS also altered the content of circulating exosomal microRNAs in obese subjects [57][58]. Also, Alkandari et al. hypothesized that BMS modulates the levels of microRNAs (miR-192 and miR-200) which was found highly expressed in urine before and after surgery in comparison to non-surgical control subjects [59]. Moreover, miR-448 and its target gene SIRT1, the latter associated with cellular metabolism, can serve as prognostic indicators for obese and T2DM patients after BMS [60]. These studies evidenced the role of microRNAs in understanding the landscape of the crosstalk between miRNAs and metabolic pathways that point to a systemic regulation of processes including multiple axes of energy metabolism. In the future, microRNAs might be useful for facilitating decisions about surgery and/or predicting weight loss after bariatric surgery, providing targets for future treatments.
3.2. Epigenetics as driving-force controlling obesity-related genes towards diabetes
Obesity and physical inactivity, probably on the background of genetic predisposition [61], are recognized as major risk factors for diabetes, and for progression of prediabetes to T2D. Longitudinal studies have shown in subjects with long-standing obesity the parallel progression from normal glucose tolerance to prediabetes and from prediabetes to diabetes. In a study on developmental trajectories of BMI from childhood into late adolescence and subsequent late adolescence–young adulthood cardiometabolic risk markers, BMI trajectory was strongly associated with HDL-cholesterol and IL-18 in males, and diastolic blood pressure and IL-6 in females. Thus, BMI trajectory was associated with cytokine levels in both sexes. This shows that BMI trajectory confers additional cardiometabolic risk beyond age-specific BMI, and suggests a strong impact of BMI trajectory on proinflammatory markers; the sex-specific trajectory–cardiometabolic risk marker association appears noteworthy [62].
FTO (fat mass and obesity-associated), a gene associated with the common forms of obesity [63] and with IR in T2D [64], modulates insulin activity and BMI [65]. FTO regulates the amount of fat deposition and affects T2D risk through its effect on BMI; indeed, people homozygous for a particular FTO allele weighed about 3 kilograms more and had a 1.6-fold greater rate of obesity than those who had not inherited this trait. At the molecular level, the driving-force for activation of proinflammatory genes is traceable to the epigenetic processes, as the chromatin arrangements, that are deputed to control gene expression in the metabolic processes and in nutritional requirements; this view would be important for stimulating structural adaptations driving to deleterious consequences on the co-morbidities of obesity, including diabetes and CVD. As other pathologies, an obesogenic environment prone towards diabetes favours the addition of methyl group to DNA, mediated by methyltransferases (DNMTs), representing the critical regulators for the stimulation of pro-inflammatory cytokines, obesity-induced, to selectively methylate and stimulate the compacting chromatin structure in the gene promoter, and thus exacerbating IR [66]. However, the methylation levels in the genes involved in lipid metabolism, such as lipoprotein lipase (LPL) essential in storing or consuming triglycerides, are altered in obese patients with the metabolic disease, at difference from healthy people [67]. In a recent work a significant influence of fats on site-specific DNA-methylation (DNA-me) relevant to T2D was found [68]; for instance, the associations between high levels of DNA-me in CpG (or CG island) site of a gene essential in cholesterol transport (ABCG1) correlated with fasting insulin and HOMA-IR in CD4+ T cells from non-diabetic individuals [69], suggesting that the derangements of the metabolic state which drive to T2D onset could be influenced by DNA-me on the obesogenic genes. Since genetic variations have occurred for the development of obesity and diabetes, genome-wide associations studies (GWA) have highlighted various genes, e.g. melanocortin-4 receptor (MC4R) gene for obesity [70], and peroxisome proliferator-activated receptor gamma (PPARG) gene [71] and islet potassium voltage-gated channel subfamily J member 11 (KCNJ11) gene [72] for T2D. More than 150 genetic loci are involved in the development of obesity and diabetes [73]. In addition, associated with adipocyte function, the gene encoding KLF14 (Krupper-Like Factor 14) appears as a key player in human metabolism being associated with IR [74][75] and the development of prediabetes. It appears more interesting that in humans hypocaloric dietary intervention changed the methylation levels of obesity-related genes in obese people [76], ameliorating the phenotype; also in mice [77] an earlier hypocaloric dietary treatment dramatically increased the DNA methylation status.
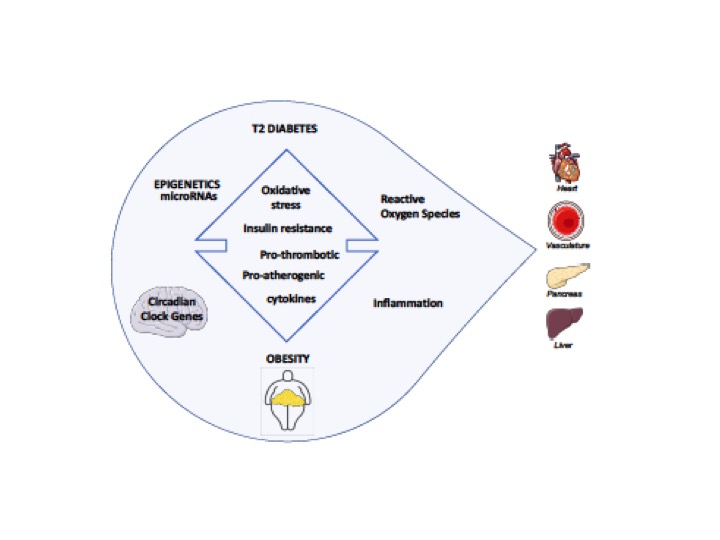
Figure 1. Major contributors of T2D/Obesity driving toward dysregulation of target tissues
References
- Collaboration, N.C.D.R.F., Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet, 2017. 390(10113): p. 2627-2642.
- Schnurr, T.M., et al., Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: a case-cohort study. Diabetologia, 2020. 63(7): p. 1324-1332.
- Prospective Studies Collaboration, Whitlock G, et al., Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet, 2009. 373(9669): p. 1083-96.
- Pontiroli, A.E., et al., Laparoscopic adjustable gastric banding for the treatment of morbid (grade 3) obesity and its metabolic complications: a three-year study. J Clin Endocrinol Metab, 2002. 87(8): p. 3555-61.
- Fuller, J.H., et al., Mortality from coronary heart disease and stroke in relation to degree of glycaemia: the Whitehall study. Br Med J (Clin Res Ed), 1983. 287(6396): p. 867-70.
- Twig, G., et al., Diabetes risk among overweight and obese metabolically healthy young adults. Diabetes Care, 2014. 37(11): p. 2989-95.
- Bjerregaard, L.G., et al., Change in Overweight from Childhood to Early Adulthood and Risk of Type 2 Diabetes. N Engl J Med, 2018. 378(14): p. 1302-1312.
- Knowler, W.C., et al., Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med, 2002. 346(6): p. 393-403.
- Lindstrom, J., et al., Sustained reduction in the incidence of type 2 diabetes by lifestyle intervention: follow-up of the Finnish Diabetes Prevention Study. Lancet, 2006. 368(9548): p. 1673-9.
- Hermanides, J., et al., Venous thrombosis is associated with hyperglycemia at diagnosis: a case-control study. J Thromb Haemost, 2009. 7(6): p. 945-9.
- La Sala, L., F. Prattichizzo, and A. Ceriello, The link between diabetes and atherosclerosis. Eur J Prev Cardiol, 2019. 26(2_suppl): p. 15-24.
- Jonk, A.M., et al., Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology (Bethesda), 2007. 22: p. 252-60.
- Avogaro, A. and S.V. de Kreutzenberg, Mechanisms of endothelial dysfunction in obesity. Clin Chim Acta, 2005. 360(1-2): p. 9-26.
- Stapleton, P.A., et al., Obesity and vascular dysfunction. Pathophysiology, 2008. 15(2): p. 79-89.
- Roden, M., et al., Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest, 1996. 97(12): p. 2859-65.
- De Nigris, V., et al., Short-term high glucose exposure impairs insulin signaling in endothelial cells. Cardiovasc Diabetol, 2015. 14: p. 114.
- Saltiel, A.R. and C.R. Kahn, Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 2001. 414(6865): p. 799-806.
- Lamia, K.A., K.F. Storch, and C.J. Weitz, Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A, 2008. 105(39): p. 15172-7.
- McHill, A.W., et al., Impact of circadian misalignment on energy metabolism during simulated nightshift work. Proc Natl Acad Sci U S A, 2014. 111(48): p. 17302-7.
- Kalsbeek, A., et al., The hypothalamic clock and its control of glucose homeostasis. Trends Endocrinol Metab, 2010. 21(7): p. 402-10.
- Turek, F.W., et al., Obesity and metabolic syndrome in circadian Clock mutant mice. Science, 2005. 308(5724): p. 1043-5.
- Shimba, S., et al., Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci U S A, 2005. 102(34): p. 12071-6.
- Rudic, R.D., et al., BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol, 2004. 2(11): p. e377.
- Kaneko, K., et al., Obesity alters circadian expressions of molecular clock genes in the brainstem. Brain Res, 2009. 1263: p. 58-68.
- Yang, Q., et al., Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature, 2005. 436(7049): p. 356-62.
- Vieira, E., et al., Altered clock gene expression in obese visceral adipose tissue is associated with metabolic syndrome. PLoS One, 2014. 9(11): p. e111678.
- Solt, L.A., et al., Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature, 2012. 485(7396): p. 62-8.
- Sitaula, S., et al., Suppression of atherosclerosis by synthetic REV-ERB agonist. Biochem Biophys Res Commun, 2015. 460(3): p. 566-71.
- La Sala, L., et al., Oscillating glucose induces microRNA-185 and impairs an efficient antioxidant response in human endothelial cells. Cardiovasc Diabetol, 2016. 15: p. 71.
- La Sala, L., et al., Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc Diabetol, 2018. 17(1): p. 105.
- La Sala, L., et al., Circulating microRNA-21 is an early predictor of ROS-mediated damage in subjects with high risk of developing diabetes and in drug-naive T2D. Cardiovasc Diabetol, 2019. 18(1): p. 18.
- La Sala, L., et al., Oscillating glucose and constant high glucose induce endoglin expression in endothelial cells: the role of oxidative stress. Acta Diabetol, 2015. 52(3): p. 505-12.
- Sharma, A., et al., The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activator dh404 protects against diabetes-induced endothelial dysfunction. Cardiovasc Diabetol, 2017. 16(1): p. 33.
- He, M., et al., Induction of HO-1 and redox signaling in endothelial cells by advanced glycation end products: a role for Nrf2 in vascular protection in diabetes. Nutr Metab Cardiovasc Dis, 2011. 21(4): p. 277-85.
- Leiter, L.A., et al., Postprandial glucose regulation: new data and new implications. Clin Ther, 2005. 27 Suppl B: p. S42-56.
- Ellulu, M.S., et al., Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci, 2017. 13(4): p. 851-863.
- Galkina, E. and K. Ley, Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol, 2009. 27: p. 165-97.
- Catalan, V., et al., Validation of endogenous control genes in human adipose tissue: relevance to obesity and obesity-associated type 2 diabetes mellitus. Horm Metab Res, 2007. 39(7): p. 495-500.
- Luft, V.C., et al., Chronic inflammation role in the obesity-diabetes association: a case-cohort study. Diabetol Metab Syndr, 2013. 5(1): p. 31.
- Hotamisligil, G.S., N.S. Shargill, and B.M. Spiegelman, Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science, 1993. 259(5091): p. 87-91.
- Gao, Z., et al., Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem, 2002. 277(50): p. 48115-21.
- Shoelson, S.E., J. Lee, and M. Yuan, Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord, 2003. 27 Suppl 3: p. S49-52.
- Green, E.A., E.E. Eynon, and R.A. Flavell, Local expression of TNFalpha in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity, 1998. 9(5): p. 733-43.
- Steppan, C.M., et al., The hormone resistin links obesity to diabetes. Nature, 2001. 409(6818): p. 307-12.
- Vandanmagsar, B., et al., The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med, 2011. 17(2): p. 179-88.
- McGeary, S.E., et al., The biochemical basis of microRNA targeting efficacy. Science, 2019. 366(6472).
- La Sala, L., et al., One-hour plasma glucose combined with skin autofluorescence identifies subjects with pre-diabetes: the DIAPASON study. BMJ Open Diabetes Res Care, 2020. 8(1).
- de Candia, P., et al., A unique plasma microRNA profile defines type 2 diabetes progression. PLoS One, 2017. 12(12): p. e0188980.
- Mysore, R., et al., MicroRNA-192* impairs adipocyte triglyceride storage. Biochim Biophys Acta, 2016. 1861(4): p. 342-51.
- Belarbi, Y., et al., MicroRNA-193b Controls Adiponectin Production in Human White Adipose Tissue. J Clin Endocrinol Metab, 2015. 100(8): p. E1084-8.
- Parrizas, M., et al., Circulating miR-192 and miR-193b are markers of prediabetes and are modulated by an exercise intervention. J Clin Endocrinol Metab, 2015. 100(3): p. E407-15.
- Jimenez-Lucena, R., et al., MiRNAs profile as biomarkers of nutritional therapy for the prevention of type 2 diabetes mellitus: From the CORDIOPREV study. Clin Nutr, 2020.
- Mingrone, G., et al., Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med, 2012. 366(17): p. 1577-85.
- Matsuyama, H. and H.I. Suzuki, Systems and Synthetic microRNA Biology: From Biogenesis to Disease Pathogenesis. Int J Mol Sci, 2019. 21(1).
- Kurylowicz, A., et al., NGS Reveals Molecular Pathways Affected by Obesity and Weight Loss-Related Changes in miRNA Levels in Adipose Tissue. Int J Mol Sci, 2017. 19(1).
- Wu, Q., et al., Metabolic phenotype-microRNA data fusion analysis of the systemic consequences of Roux-en-Y gastric bypass surgery. Int J Obes (Lond), 2015. 39(7): p. 1126-34.
- Hubal, M.J., et al., Circulating adipocyte-derived exosomal MicroRNAs associated with decreased insulin resistance after gastric bypass. Obesity (Silver Spring), 2017. 25(1): p. 102-110.
- Bae, Y.U., et al., Bariatric Surgery Alters microRNA Content of Circulating Exosomes in Patients with Obesity. Obesity (Silver Spring), 2019. 27(2): p. 264-271.
- Alkandari, A., et al., Bariatric Surgery Modulates Urinary Levels of MicroRNAs Involved in the Regulation of Renal Function. Front Endocrinol (Lausanne), 2019. 10: p. 319.
- Wang, Y., et al., Expression of MicroRNA-448 and SIRT1 and Prognosis of Obese Type 2 Diabetic Mellitus Patients After Laparoscopic Bariatric Surgery. Cell Physiol Biochem, 2018. 45(3): p. 935-950.
- Said, M.A., N. Verweij, and P. van der Harst, Associations of Combined Genetic and Lifestyle Risks With Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA Cardiol, 2018. 3(8): p. 693-702.
- Oluwagbemigun, K., et al., Developmental trajectories of body mass index from childhood into late adolescence and subsequent late adolescence-young adulthood cardiometabolic risk markers. Cardiovasc Diabetol, 2019. 18(1): p. 9.
- Frayling, T.M., et al., A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science, 2007. 316(5826): p. 889-94.
- Zeggini, E., et al., Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science, 2007. 316(5829): p. 1336-41.
- Frayling, T.M., Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet, 2007. 8(9): p. 657-62.
- Kim, A.Y., et al., Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat Commun, 2015. 6: p. 7585.
- Castellano-Castillo, D., et al., Altered Adipose Tissue DNA Methylation Status in Metabolic Syndrome: Relationships Between Global DNA Methylation and Specific Methylation at Adipogenic, Lipid Metabolism and Inflammatory Candidate Genes and Metabolic Variables. J Clin Med, 2019. 8(1).
- Dick, K.J., et al., DNA methylation and body-mass index: a genome-wide analysis. Lancet, 2014. 383(9933): p. 1990-8.
- Hidalgo, B., et al., Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network study. Diabetes, 2014. 63(2): p. 801-7.
- Larsen, L.H., et al., Prevalence of mutations and functional analyses of melanocortin 4 receptor variants identified among 750 men with juvenile-onset obesity. J Clin Endocrinol Metab, 2005. 90(1): p. 219-24.
- Altshuler, D., et al., The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet, 2000. 26(1): p. 76-80.
- Gloyn, A.L., et al., Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes, 2003. 52(2): p. 568-72.
- McCarthy, M.I., Genomics, type 2 diabetes, and obesity. N Engl J Med, 2010. 363(24): p. 2339-50.
- Voight, B.F., et al., Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet, 2010. 42(7): p. 579-89.
- Small, K.S., et al., Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet, 2011. 43(6): p. 561-4.
- Nicoletti, C.F., et al., DNA methylation pattern changes following a short-term hypocaloric diet in women with obesity. Eur J Clin Nutr, 2020. 74(9): p. 1345-1353.
- Sziraki, A., A. Tyshkovskiy, and V.N. Gladyshev, Global remodeling of the mouse DNA methylome during aging and in response to calorie restriction. Aging Cell, 2018. 17(3): p. e12738.

