 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Krzysztof Gwozdzinski | + 6623 word(s) | 6623 | 2020-11-04 09:29:37 |
Video Upload Options
Venous thromboembolism (VTE) refers to deep vein thrombosis (DVT), whose consequence may be a pulmonary embolism (PE). Thrombosis is associated with significant morbidity and mortality and is the third most common cardiovascular disease after myocardial infarction and stroke. DVT is associated with the formation of a blood clot in a deep vein in the body. Thrombosis promotes slowed blood flow, hypoxia, cell activation, and the associated release of many active substances involved in blood clot formation. All thrombi which adhere to endothelium consist of fibrin, platelets, and trapped red and white blood cells.
1. Introduction
Thrombosis can occur in either veins or arteries. In the veins, it leads to DVT and a pulmonary embolism, referred to as VTE. In turn, in the arteries, it most often causes myocardial infarction, ischemic stroke, and acute limb ischemia [1]. The term DVT usually refers to a blood clot that forms in the deep vein, most often in the large veins that calf muscles and eventually leads to pain and swelling in the leg. If the thrombosis is not treated, a pulmonary embolism may occur [2].
Venous thrombosis is becoming more common and is associated with our lifestyle. It can occur very quickly within an hour or over a long period with worsening symptoms. Long hours of travel, e.g., by plane or car, contribute to thrombosis. Moreover, hormonal therapy, including contraception, is also a contributing factor. Other factors conducive to the development of thrombosis can also include surgery, immobilization as a consequence of a procedure or caused by a long-term treatment, delivery, infectious diseases, pre-existing vein diseases, excessive coagulation, high haematocrit (increase blood viscosity), the use of drugs that irritate the endothelium, and varicose veins [3].
Thrombosis may also occur in pregnant and postpartum women. It affects not only people sitting motionless but may occur after intensive effort as well, and it can also occur after dehydration of the body. It also applies to patients undergoing an orthopaedic surgery. Diabetes, hypertension, but also smoking and being overweight, are additional risk factors for thrombosis [4].
Other factors favouring thrombosis are the age of the patients, a sedentary or standing lifestyle, lack of physical activity, obesity, as well as existing cardiovascular diseases such as hypercoagulability of the blood and the presence of varicose vein and recent surgery but also rheumatism, diabetes, smoking, and others [5][6]. Cases of thrombosis are also associated with intravenous drug users (IDUs) in whom 22% of DVT cases were reported. It is estimated that the number of drug users in the world ranges from 11 to 21 million including 4–47.6% of people with DVT [7].
DVT is associated with the formation of a blood clot in a deep vein in the body. However, blood clots can occur most often in the large veins of the calf and thigh muscles. Depending on the size, a blood clot that forms in a vein can narrow the vessel or close it completely. The consequence is the appearance of pain and swelling in the leg, as well as of serious complications such as a pulmonary embolism when a blood clot travels to the lungs leading to the inhibition of blood flow [5].
The clots can also break away and be transferred from the flowing blood to the cerebral vessels leading to an ischemic stroke. Therefore, it is very important to quickly diagnose DVT and immediately start its treatment [2]. Essential in the formation of thrombosis are endothelial cells (ECs), which provide many haemostatic regulatory molecules. The normal endothelium has anticoagulant and antithrombotic properties. Damage to the vessel causes adhesion of the platelets with the participation of the von Willebrand factor (vWF) secreted by the cells. ECs also produce tissue plasminogen activator (t-PA), its inhibitor, and an inhibitor of the tissue factor (TF) pathway. The endothelial surface is additionally the site of thrombin inactivation by antithrombin (AT) and conversion of prothrombotic thrombin by thrombomodulin into anticoagulant protein. In addition, thrombomodulin cooperates with protein C, and the thrombin-activated fibrinolysis inhibitor maintains the endothelial microenvironment in an anti-inflammatory and anticoagulant state [8][9]. Additionally, platelet activation performs a pivotal role in both infection-induced thrombosis and the immune response against pathogens.
Common symptoms of venous thrombosis include swelling, red and tense skin, sometimes a bluish tinge, and an increase in temperature in the extremity. Other symptoms are pain in the foot, calf, and lower knee, which disappears when the limb is lifted. However, one of the dangers of this serious disease is that in many cases it develops asymptomatically, and the patient is unaware of thrombosis. This is the case when the clot is attached to the wall of the vessel and does not completely close the vessel lumen, and some of the flowing blood is taken over by the surrounding smaller vessels. In addition, the blood clot may shrink, which results in an increased blood flow. In the best situation, the blood clot dissolves, and the vessel is restored. Unfortunately, endogenous protective and repair mechanisms are not always sufficient, and the formation of blood clots often leads to complications.
2. The Basic Mechanism of Blood Clotting
This section presents blood coagulation pathways and the participation of various coagulation factors in haemostasis. Discussing this topic is important as the rest of the work will present drug-inhibitors of pathways and coagulation factors in thrombosis. Blood clotting is a natural physiological process that prevents blood loss in the case of a damaged vessel. The blood clotting process is one of the body's defences when the continuity of the vascular bed is interrupted. The basis of the blood coagulation process is the formation of a clot from fibrin with the participation of thrombin [10].
Thrombin, an important molecule in the clotting process, cleaves fibrinogen, transforming it into fibrin, which is the main component of the clot. In a further stage, fibrin is cross-linked with the active FXIII to a form resistant to the action of fibrin-degrading enzymes (Figure 1) [11].
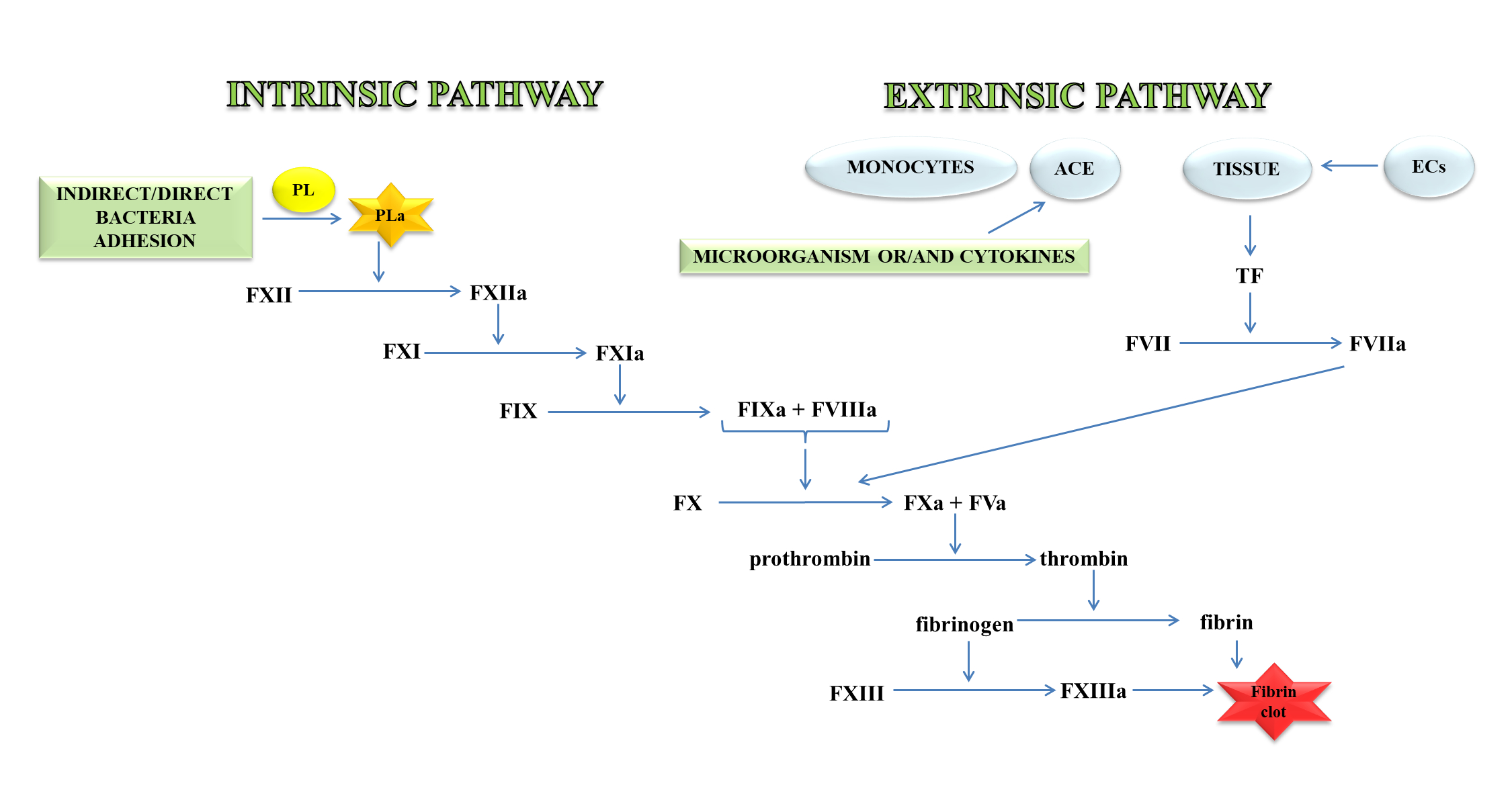
Figure 1. A blood coagulation cascade including the internal and external paths. The former of these pathways is initiated by the contact of FXII with a negatively charged surface or indirectly through the involvement of polyP and blood platelets (PL). In contrast, the external pathway is initiated by FVII activation by tissue factor (TF) on the surface of monocytes, microparticles, activated endothelial cells (ECs), or cells in the damaged vessel wall, which exhibit TF. Both pathways lead to the activation of FX factor (prothrombinase), which converts inactive prothrombin into thrombin. The next stage is the transformation of fibrinogen to fibrin, which forms a fibrin clot; abbreviation: activated platelets (PL-a), angiotensin-converting enzyme (ACE).
The external mechanisms are associated with TF, initiated by tissues or monocytes. TF is an integral type I membrane protein (glycoprotein). A fragment of this glycoprotein is exposed on the membrane surface and binds with very high affinity to FVII. Complex TF-FVII is rapidly converted by limited proteolysis to the activated form of FVII [12]. The internal mechanism occurs due to the action of FXII with the surface having a negative charge. Both mechanisms lead to the activation of the enzymatic transformation cascade FX. The spatial network of a clot is stabilised by transglutaminase, FXIII. In addition, damage to the vessel leads to the discovery of the subendothelial layer, to which the platelets of blood immediately adhere that, as a result of activation, form a clot, inhibiting bleeding from a damaged arteriole, small vein or capillaries. The formation of the clot is caused by the presence of adhesive proteins that participate in the bonding of the tiles between them. The most important protein involved in this process is the platelet integrin αIIbβ3 also called the glycoprotein (GP) IIb/IIIa receptor. In the blood coagulation cascade, other factors such as FVII, FIX, FX, FXI, FXII and activated forms are also involved: FVa, FVIIa, FVIIIa, FIXa, Fxa, FXIa, FXIIa, FXIIIa, and PolyP [13]. Briefly, the process of blood clotting under conditions of haemostasis has been described, taking into consideration the internal and external pathways and coagulation factors.
3. Causes of Vein Thrombosis
Already more than 100 years ago, Rudolf Virchow stressed that the cause of thrombosis is the damage to the sinus, endothelial damage, coagulation disorders that lead to an increase in blood viscosity and slow its flow. Arterial thrombosis occurs under conditions of severe shear and is associated with platelets. In contrast, venous thrombosis occurs with low shear flow and mainly around the intact endothelial wall. Arterial thrombosis is associated with the formation of platelet-rich clots in the arteries as a result of atherosclerotic plaque rupture. The detachment of the thrombus can lead to a heart attack, stroke, transient ischaemic attack and limb and intestinal ischemia. On the other hand, thrombosis is related to the formation of clots in the veins of blood cells, low in platelets but rich in fibrin and plasma clotting factors. In thrombosis under hypoxic conditions, a thrombus forms, which causes increased blood clotting activity. Unlike arterial thrombosis, thrombosis causes ischemia at the site of origin. Thrombosis is promoted by the presence of cancer, especially lung and pancreatic cancers, which is associated with disturbances in homeostasis and the clotting process [14].
Blood flow in deep veins is very slow and, hence, the most common thrombosis occurs in these vessels, where the main mechanism forcing blood flow is the spasm of the shins. However, this mechanism stops working when the muscles do not shrink for a long time. Such a phenomenon occurs most often after surgery, in diseases leading to being bedridden, and in people with lower-limb paralysis. The frequency of complications is higher in patients undergoing an orthopaedic surgery than in others; this applies to, for example, a hip fracture. DVT in operated people is initiated by several of factors, such as the release of TF in the operative field, the release and absorption of thrombin from the surgical wound, and imbalance in the fibrinolytic system [15]. Besides, intravenous administration of general anaesthetics leads to a decrease in blood flow. In turn, medications from the curare or succinylcholine group that cause immediate muscle relaxation, reduce the effect of the muscle on the venous vessels and lead to a slowdown in blood flow to a minimum [16].
In most patients, sepsis also leads to the activation of the coagulation system and, consequently, thrombosis. Inflammation in the vein wall initiates the formation of a thrombus in a normal vessel. The first stage of clot formation is the activation of ECs, platelets, and leukocytes associated with the induction of inflammation and the release of microparticles that trigger the coagulation system by induction of the TF [17][18]. The thrombus is formed when platelets and leukocytes adhere to the vessel wall, and it is further promoted by inflammation and tissue factor as well as proinflammatory cytokines that activate coagulation. Leukocyte adhesion occurs when P-selectin glycoprotein ligand-1 (PSGL-1) present on the surface binds P-selectin expressed on activated endothelial cells. When P-selectin is also exposed to the surface of blood platelets, it binds to leukocytes leading to inflammation. Furthermore, exposure of P-selectin to the surface of platelets leads to rapid exposure of TF in the blood through monocytes contributing to intravascular coagulation [19].
Thrombosis is not always associated with the occurrence of complications. Occasionally, the thrombus is partially or completely dissolved in the vessel. Sometimes this process may last longer before it is completely dissolved, displaying practically no symptoms. During this period, part of the function related to the blood flow will be taken over by the smaller vessels. DVT has typical signs and symptoms, including pain, swelling, heat, redness or discoloration, and distention of surface veins. However, about half of people with this disease have no symptoms [20]. The presence of a clot can be diagnosed with an X-ray or less invasive methods such as conventional contrast venography, ultrasound, computed tomography (CT), or magnetic resonance imaging (MRI) scans [5].
4. Platelets and endothelial cells activation
Under conditions of haemostasis, a balance is maintained through the interaction between platelets and vascular endothelium, which is also controlled by the cascade of roots and fibrinolysis [21]. Platelets play a crucial role in hemostasis and thrombosis, and their interactions with the endothelium are important in cardiovascular diseases, including atherosclerosis and thrombosis. Endothelial dysfunction and platelet hyperreactivity lead to an increased risk of these diseases.
Activated platelets (PLa) release a number of active substances from the a and b granules, which additionally intensify their activation. The platelets also liberate serotonin, which causes narrowing of the blood vessels in the wound [13]. In addition, stimulated platelets release other adhesive GPs from granules such as fibrinogen; thrombospondin; vWF; coagulation factors: FV, FXI, FXIII, protein S; mitogenic factors: platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), epidermal growth factor (EGF); angiogenic factors: vascular endothelial growth factor (VEGF), PF4 inhibitor; chemokines: chemokine CXC ligand (CXCL7), CXCL4, CHCL1, CXCL5, CCL5 (RANTES), CCL3 and cytokines: IL-1 β, TGF-β, fibrinolysis inhibitors, plasminogen activator inhibitor-1 (PAI-I) [22].
Numerous experimental and clinical studies confirm the key role of ROS in the mechanism of platelet activation. There are many activation pathways, one of which is an increased isoprostane formation. The main source of superoxide and other ROS is NADPH oxidase 2 (NOX2) [23]. One source is the arachidonic acid (AA) metabolism and associated lipoxygenase (LOX) expression, the other is NADPH oxidase (NOX) [24]. ROS form within PLa and regulate their responses to collagen-mediatedthrombus formation [25].
Additionally, inflammation initiated by pathogens also leads to the activation of blood platelets, if accompanied by damage to the endothelium, then fibrin builds up and a clot forms [26]. Bacteria can also release substances that activate platelets [27]. Both infection and inflammation can directly or indirectly activate platelets through different receptors, triggering aggregation and thrombi formation within the vasculature. However, many reviews based on current literature summarize the impact of potential risk factors related to the involvement of platelets in the pathogenesis of VTE, as well as in predicting the risk of VTE events [28]. Additional risk factors may be expression markers such as tissue factor containing microparticles and P-selectin, which is a platelet activation marker, enhances microparticle production, tissue factor expression, and platelet aggregation [29].
Endothelial cells have a key role in homeostasis by having a direct contact between the blood and the vessel wall. They are involved in various functions such as regulation of blood flow, vascular permeability, recruitment of inflammatory cells, thrombosis, and angiogenesis. These cells secrete molecules that act on smooth muscle cells associated with vascular tone regulation and leukocytes that regulate inflammation state. ECs release prostacycline (PGI) and NO•, both substances that inhibit platelet adhesion and vasodilators (Figure 2) [8][30].
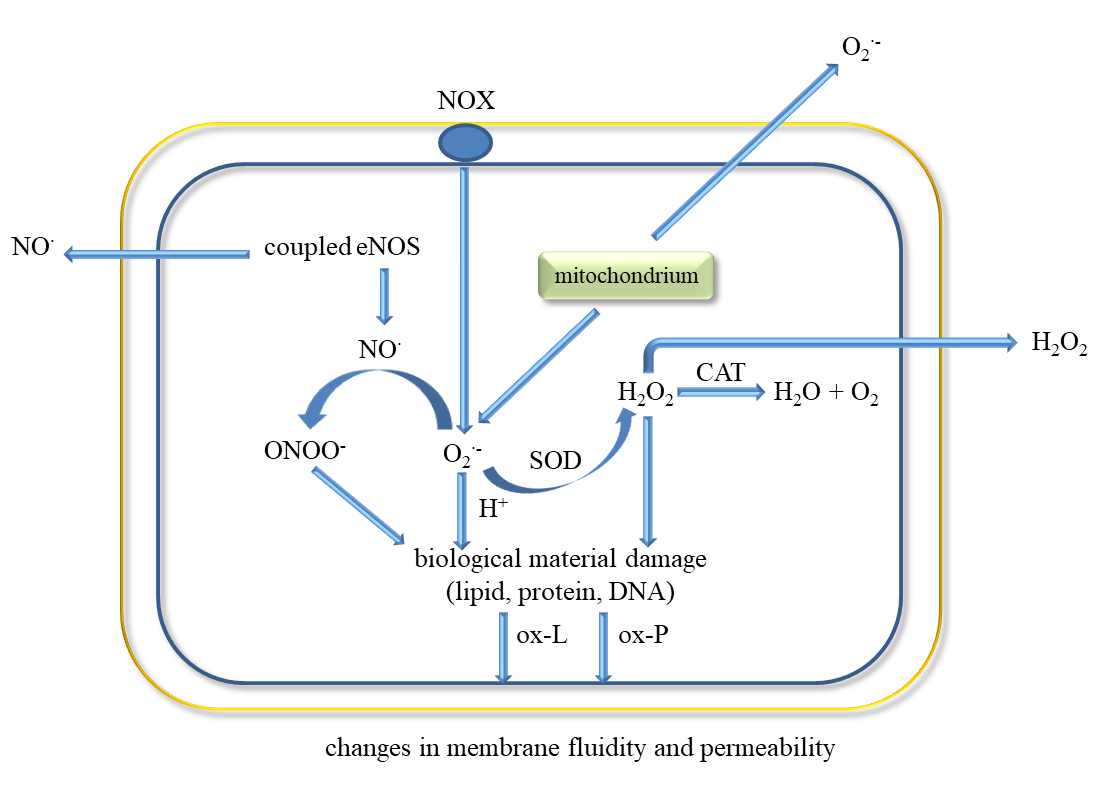
The classic factors leading to endothelium dysfunction include dyslipidaemia, smoking, hypertension diabetes mellitus, and aging. Other factors are infection inflammation, physical inactivity, high level of homocysteine, post prandial state, and obesity [30]. Excessive generation of ROS in the vascular wall is a common feature of many cardiovascular diseases. Endothelial dysfunction occurs in both varicose veins and DVT [31]. It has been shown that ECs can be damaged by the endotoxin part of outer membrane of Gram-negative bacteria in bacterial pathogenesis and endotoxic shock [32]. The factors leading to the development of endothelial dysfunction also include ROS, pro-inflammatory cytokines (IL-1, IL-6, IF-8, TNF-α), TGF-β, Hb and oxyHb, Th-1 lymphocytes, sepsis, macrophages and neutrophils.
Activation of ECs is associated with increased expression of cellular adhesion molecules VCAM-1 and ICAM-1, which causes ED and promotes monocyte adhesion [33]. This process can be triggered by ischemia, catecholamines, angiotensin II, cytokines (IL-1, IL-6, TNF-α, TGF-β), and endotoxins [34][35][36].
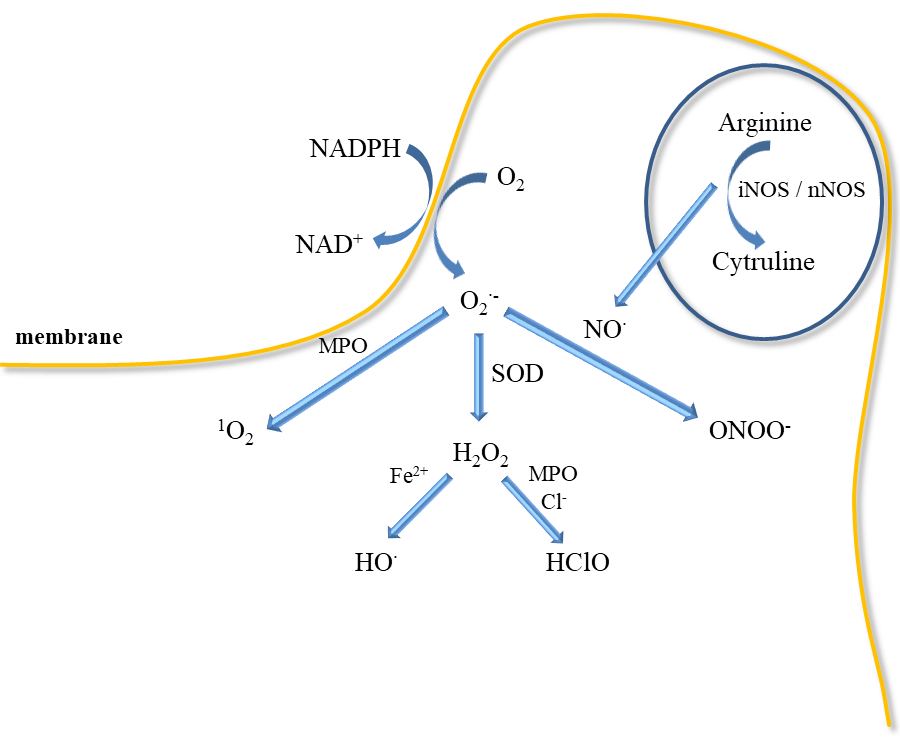
Inflammatory mediators promote the adherence of neutrophils to the endothelium and their activation. Activated neutrophils, being inflammatory cells, as a result of respiratory burst release large amounts of ROS such as superoxide, H2O2, hydroxyl radical (HO•), nitric oxide, and hypochlorous acid (HClO) (Figure 3). ROS produced during inflammation lead to oxidative stress but are also used to remove pathogens [37]. In addition, ED leads to increased production of O2•− and H2O2 catalysed by xanthine oxidase, which is accompanied by an increase in the synthesis of peroxynitrite, the factor responsible for damaging the biological material [38]. Activation of endothelial cells releases many active substances that may be involved in clot formation.
5. ROS and Oxidative Stress
The release of lower ROS levels by host cells is associated with their physiological function in regulating processes such as haemostasis, apoptosis, immune response, and microbial colonisation [39]. ROS are produced of numerous enzymatic reactions in various cell compartments, including the cytoplasm, cell membrane, endoplasmic reticulum, mitochondria, and reactions peroxisome as intra- and intercellular signalling molecules. The precursor of ROS is the superoxide anion radical (O2•−), which is formed as a result of a one-electron reduction of molecular oxygen. Superoxide anion radical is formed by the enzymatic process, autooxidation reaction, and by nonenzymatic electron transfer reactions in which an electron is transferred to molecular oxygen [40]. Superoxide is mostly produced within the mitochondria in electron transport chain during electron leakage. There are 11 sites in the respiratory chain in mammalian mitochondria where superoxide and/or hydrogen peroxide is formed [41]. Other enzymes that can produce superoxide are lipoxygenase, cyclooxygenase [42]. Superoxide can exist in two forms such as O2•− or hydroperoxyl radical (HO2•) at low pH [43]. Its reactivity with the biomolecules is low while the reactivity of hydroperoxyl radical is much higher. Hydroperoxyl radical can penetrate the phospholipid bilayer easier than the charged form (O2•−). Polymorphonuclear leukocytes (PMNs), monocytes, and macrophages perform an important role in the innate immune response to pathogens. They can produce and release ROS in significant amounts [44]. One of the main sources of peroxide in phagocytic cells is membrane NAD(P)H oxidase [45]. Spontaneous or enzymatic dismutation of superoxide leads to hydrogen peroxide. Hydrogen peroxide can diffuse through the mitochondrial membranes into the cytoplasm [46]. The superoxide produced during phagocytosis is transformed into other ROS, which are directed to the absorbed material in the phagolysosome. Depending on the way used to activate the NAD(P)H oxidase, the production of superoxide can also target the extracellular environment [47]. ROS perform a physiological role as signalling molecules; however, their increased production and/or failure of antioxidative systems may lead to oxidative stress, which is associated with damage/modification of life-important molecules such as DNA, proteins, and lipids. It has recently been shown that certain molecules produced during the blood clotting process can stimulate PMNs to produce ROS. Released ROS by PMNs can damage the endothelium and affect the coagulation process [48].
The release of high ROS levels due to acute infection or inflammation may lead to oxidative damage to the host's biological material. ROS have a crucial role in vascular function. Redox balance is important in cell signalling and haemostasis. However, perturbation of redox state leads to ROS interaction with important vital macromolecules such as proteins, enzymes, nucleic acids that can lead to vascular pathology [49][50]. ROS are associated with the pathogenesis of many diseases, including cardiovascular diseases such as hypertension, ischemic heart disease, ischemia-reperfusion injury, and other vascular diseases including thrombosis. ROS may also lead to other pathological conditions, such as pulmonary fibrosis and vascular retinopathy [34][51].
In pathological conditions, there is a continuous recruitment of inflammatory cells that release ROS [52]. This process is observed, for example, in atherosclerosis and a long-term venous disease. Excessive ROS formation and/or a decrease of antioxidant potential leads to changes in the vessel wall [53]. Eukaryotic cells have numerous organelles in which ROS can be produced. The largest amounts of ROS are released in the ETC in the mitochondria [54].
In the vascular system, ROS are generated mainly by NOX but also uncoupled NOS, xanthine oxidase, COX, and myeloperoxidase (MPO). NOX occurs in the isoforms NOX1, NOX2, NOX4, and NOX5. It was shown that the expression of isoforms 1, 2, 4 is the cause of cardiovascular diseases [51][55][56]. NOX4 participates in the production of oxygen active compounds through internal mitochondrial channels, including ATP-dependent potassium channels and mitochondrial permeability transition pore (mPTP) through which ROS can flow from the mitochondria to the cytosol [57][58]. NOX4 induced by TGF-β lead to ROS production, inhibition of mitogen-activated protein kinases (MAPK), MKP-1, and activation of RhoA [59]. NOX4 expression leads to DNA damage by H2O2, which was also shown diffused through nuclei membrane [60]. NOX can be enhanced by exogenous factors such as ultraviolet (UV) radiation, smoking, high calorie diets, and diabetes [51]. All these enzymes are complexes present in the membrane and are expressed in vascular tissue only to produce ROS. For example, NOX2 and NOX4 produce ROS in cardiomyocytes and fibroblasts, while NOX1, NOX4, and NOX5 do so in vascular smooth muscle cells (VSMCs) [59]. Other sources of ROS include peroxisomes, endoplasmic reticulum, and lysosomes, where they are produced by NOX, xanthine oxidases, cytochrome P450 (CYP), etc. [61].
However, large amounts of ROS are released by neutrophils during respiratory burst in response to infection because they have a crucial role in host defence and inflammation (Figure 3). Neutrophils participate in the immune responses associated with bacterial invasion, and they also produce nitric oxide, which is synthesised by neutrophilic inducible nitric oxide synthase. In addition to various physiological functions, their participation is associated with pathology [62]. Neutrophils may participate in numerous pathologies such as glomerulonephritis, diabetes, stroke, septic shock, whooping cough, sepsis, encephalitis, and ulcerative colitis. NO• has been shown to mediate neutrophil extracellular traps (NETs) formation at the site of inflammation/infection by increasing ROS production. Activated neutrophils can form NETs, which include proteins, histones, chromatin, and DNA [41]. In turn, NETs increase coagulation by stimulating platelet aggregation, erythrocyte mobilization, and fibrin accumulation as well as endothelial activation and damage [63][64]. NO• is also a mediator of neutrophil response in a neurodegenerative disease (Parkinson's disease, schizophrenia, depression) and cardiovascular diseases, thrombosis, and hypertension [62][65]. Higher iNOS activity in neutrophils was observed in patients with congestive heart failure that positively correlated with elevated levels of IL-6 and noradrenaline in circulation [66].
The mechanism of bacteria removal by ROS is associated with an attack on membranes, but also with their penetration into the interior of bacteria, where they can damage nucleic acids, proteins, and enzymes [67]. However, NOX function is crucial for the formation of NETs [68]. NETs formation is also induced by proteases, which can be activated by ROS. Neutrophils can generate more ROS by responding to AGEs through the NOX complex [69][70]. Additionally, superoxide may be also released in mitochondria by NAD(P)H-oxidase, cytochrome c peroxidase or xanthine oxidase (Figure 3). Myeloperoxidase located in neutrophil granules can, with the participation of H2O2, oxidise chlorides to HClO, which is a strong oxidising agent and bacteria killing agent. MPO can also directly convert superoxide to singlet oxygen (1O2). Singlet oxygen can also be produced in a reaction of hypochlorite with hydrogen peroxide. However, the released H2O2 can be reduced in the presence of iron II to the HO• (Fenton reaction). In addition, both ROS stimulate the production of proinflammatory cytokines as TNF-α and macrophage inflammatory protein 2 (MIP-2) [67][68][69].
Another important source of ROS in platelets are NOX occurring in various cell types, including ECs and other vascular cells. NOX expression can be triggered by hypoxia and endogenous factors, such as pro-inflammatory molecules, growth factors, PDGF, and hormones: angiotensin II and insulin [71][72]. ROS are also generated in the cells of VSMCs. This mechanism involves extracellular cyclophilin A (iCypA), which increases ROS production by angiotensin II [73].
Oxidative stress initiates the NF-κB transcription factor, which can cause the expression of AGEs and TNF-α [74]. Additionally, AGEs may promote oxidative stress which leads to the formation of oxidative protein products [75]. TNF-α is a pro-inflammatory cytokine that is released by active monocytes and macrophages and is associated with the inflammatory process in the host's response to infection [76].
Proper functioning of the endothelium is also associated with the release of NO• by endothelial nitric oxide synthase (eNOS) in which calcium ions participate [74]. Nitric oxide characterised by a low reactivity, free of electric charge, easily penetrates through the membranes of ECs and can be oxidized to nitric dioxide [77]. In turn, the reaction of NO• and the O2•− leads to formation of ONOO-, which is characterised by similar properties as nitrogen dioxide, both inflammatory agents [78][79]. The constant rate of reaction of NO• with O2•− is about three times higher than dismutation of superoxide by superoxide dismutase (SOD) [80][81][82]. This supports the thesis that higher levels of superoxide and nitric oxide are harmful and in pathology lead to a damage to biological material [83]. In turn, oxyHb released in plasma can react with NO• and produce ONOO− and metHb [84][85]. It was shown that treatment of RBCs with nitric oxide led to metHb formation and oxidative damage of lipids and proteins in these cells [86]. It was reported that the lipid peroxidation product, 4-hydroxy-2-nonenal (HNE), causes changes in cryptic TF on the surface of blood macrophages and monocytic cell line, THP-1, converting it to TF procoagulant [87].
ROS affecting the processes of coagulation, fibrinolysis, proteolysis and complement, as well as the activation of platelets, endothelial cells, erythrocytes, neutrophils, monocytes may lead to the formation of a venous thrombus [88]. Furthermore, ROS can initiate coagulation inducing increased expression of TF in endothelial cells, monocytes, and vascular smooth muscle cells, but NOX enzymes are necessary for ROS production [89][90][91]. It has recently been shown that certain molecules produced during the blood clotting process can stimulate PMNs to produce ROS. Released ROS by PMNs can damage the endothelium and affect the coagulation process [48].
6. Advances in Treatment of Thromboembolism
Conservative therapy is based on the administration of anticoagulants that reduce the risk of a pulmonary embolism. Anticoagulants have become the basis of DVT therapy and are used to prevent PE progression and recurrence of thrombosis. It was reported than the 30-day mortality risk for VTE patients was 3% for DVT not treated with anticoagulants. However, mortality risk increases to 31% in patients who have developed PE. In addition, anticoagulants inhibit the growth of existing thrombi and prevent the emergence of new ones. These medicines include low molecular weight heparin (LMWH), acenocoumarol, aspirin, and other antiplatelet inhibitors [92]. In addition, natural phlebotropic drugs such as rutin, aescin, pycnogenol, Ruscus or Centella extracts, Ginkgo biloba, and others strengthen and protect the walls of venous vessels [93].
The therapy uses oral anticoagulants for 3–6 months. Discontinuation of treatment in patients after the first episode of idiopathic VTE causes recurrent thrombosis with a frequency of approximately 5–10% in the first 2 years and in the following 1–3%. On the other hand, the long-term use of anticoagulants is associated with haemorrhagic complications. It is very important to clarify in which patients there is an increased risk of VTE recurrence. It is connected with the decision to extend the therapy and the isolation of the group of patients with low risk of VTE relapse, in whom anticoagulant therapy can be safely discontinued [94].
The variety of anticoagulants is associated with their use for the treatment and prevention different disease such as atrial fibrillation, deep vein thrombosis, pulmonary embolism, Ischemic stroke, myocardial infarction (heart attack), hip or knee replacement surgery, and others. Some drugs do not dissolve thrombi but prevent the growth of those already formed and the formation of new ones. In contrast, another group of drugs called thrombolytics ("clot busters") can dissolve a clot during a period of a few days.
Drugs used to treat or prevent thrombosis can be divided into three groups: (1) anticoagulants that prevent the coagulation system and interfere with further plaque expansion, (2) antiplatelet agents that reduce platelet aggregation and inhibit blood clot formation, (3) fibrinolytic drugs that directly dissolve the resulting clot [95].
In thromboembolic syndromes, coagulation FXa has a key role in the blood coagulation cascade. Factor Xa participates in both the external and internal coagulation pathways and is important in conversion of prothrombin to thrombin [96][97]. Direct FXa inhibitors may include: apixaban, betrixaban, darexaban and other drugs. In turn, indirect FXa inhibitors: fondaparinux, idraparinux, dabigatran and LMWH. Both group of FXa inhibitors are used to reduce the risk of stroke and embolism in people with nonvalvular atrial fibrillation. They are also used to prevent DVT, which can lead to a pulmonary embolism (PE) in patients after a knee or hip surgery, as well as to treat DVT and PE, and to reduce the risk of both diseases coming back [98][99][100].
Another group of drugs involved in the inhibition of thrombus formation are thrombin inhibitors, such as unfractionated heparin (UFH), LMWH, bivalirudin, and hirudin. Thrombin activates FV, FVIII, and FXI, which participate in the release of more thrombin and activate FXIII, a protein involved in fibrin cross-linking and clot stabilisation [101]. In thrombosis, pathological activation of platelets occurs, which in turn leads to uncontrolled clot growth, embolism, or blockage of the blood vessel. The corollary of this is organ ischemia [102][103]. The most commonly used antiplatelet agents include acetylsalicylic acid (ASA) and clopidogrel as well as dipyridamole and cilostazol as phosphodiesterase inhibitors [3][104][105]. In the treatment of thrombosis, antiplatelet drugs are used, which include: COX-1 inhibitors, adenosine diphosphate (ADP) P2Y12 receptor antagonists, GP IIb/IIIa inhibitors, GP VI, GP IB/IX/V, prostaglandin E (PGE) synthase inhibitors, serotonin receptor 2A (5HT2A), PAR, and TXA2 inhibitors [102][103]. Clopidogrel, prasugrel, and ticlopidine are active antiplatelet drugs, irreversible, competitive, thienopyridine P2Y12 receptor antagonists. In contrast, ticagrelor, cangrelor and selatogrel belong to the competitive reversible P2Y12 receptor blockade group [106][107].
In the process of haemostasis and thrombosis, the PI3Kβ/protein kinase B (Akt) signalling pathway associated with platelet activation and aggregation is important. The family of phosphoinositide 3-kinases (PI3Ks), which catalyse the phosphorylation of the inositol ring of phosphatidylinositol, acts as regulator of cellular function supporting platelet activation and thrombus formation. PI3K enzymes are also involved in cardiovascular diseases including angiogenesis, hypertension, and heart failure, and are the target of promising treatment for the prevention of thrombosis [108].
TGX221 related pyrido[1, 2-a]pyrimidin-4-one belongs to a new target for antithrombotic therapy, and it is a PI3Kβ inhibitor. In platelets, PI3Kβ is an effector of various cell surface receptors including GP Ib, GP VI, and P2Y12 [109]. The TGX-221 p110b inhibitor blocked IGF-1 initiated phosphorylation of Akt in platelets deficient in p110α, indicating that both p110α and p110β were involved in IGF-1 mediated phosphorylation of Akt [110]. Additionally, TGX221 has a particular role in shear stress conditions, becoming a new anticoagulant drug aimed at PI3Kβ [111]. In vivo studies have shown that inhibition of PI3Kβ provides protection against arterial thrombosis, with a limited effect on normal haemostasis [112].
During trauma and/or high shear stress, the GP Ib/IX/V receptor, which binds to vWF, has a key role, resulting in platelet adhesion to the endothelium. The drug that binds the A1 domain of vWF is aptamer ARC1779, 40-mer modified DNA/RNA oligonucleotide. Additionally, ARC1779 is the ligand for GP Ib receptor on platelets [113][114]. For example eptifibatide is characterised by its antithrombotic activity, which selectively and reversibly binds to the platelet GP IIb/IIIa receptors by blocking the binding of fibrinogen, vWF and other adhesive molecules [115]. A promising drug seems to be the humanised, bivalent Caplacizumab nanobody (ALX-0081) that binds to GP Iba on vWF. ALX-0081 has been approved by FDA in the treatment of adult patients with TTP [116][117]. Another vWF-binding drug, which is also a GP Iba antagonist, is the snake venom anfibatide of Agkistrodon acutus, which inhibits the interaction between vWF and PLa and consequently blocks platelet adhesion and aggregation [118]. In turn, the tripeptide of the isolated venom Agkistrodon acutus successfully inhibited the binding of fibrinogen to GP IIb/IIIa [119]. In addition, anfibatide had also a protective effect on cerebral ischaemia and reperfusion injury and its use could be beneficial for the treatment of ischaemic stroke [120][121]. A new strategy in the treatment of thrombosis is to use FXI and FXII inhibitors. Both factors are important in thrombosis but not in haemostasis. Factor FXI has a much more important role in thrombosis than FXII. In addition, the anti-FXI antibody inhibits the activation of platelets and fibrin deposition more than in the case of FXII [122].
7. Natural Compounds
There is an abundance of drugs used in thrombosis; however, they lead to non-desirable side effects, mainly bleeding. The increase in cardiovascular incidents requires the introduction of new drugs that would be effective in the treatment and prevention of thrombosis and at the same time be free of side effects. Many plant compounds such as saponins, flavonoids, and others have an important role in preventing various diseases. Oxidative stress promotes thrombotic complications. Therefore, various substances of natural origin reduce the level of oxidative stress and may be important in the treatment of thrombotic complications [123]. Inactivation of ROS and alleviation of oxidative stress can reduce the risk of platelet hyperactivation, which leads to cardiovascular diseases including thrombosis [124]. Numerous works have shown that compounds of natural origin can reduce the risk of cardiovascular diseases including thrombosis. In Far Eastern medicine, medicines of vegetable origin have been used that have good healing properties and no side effects. These substances affect the mechanisms of thrombosis in various ways, affecting the coagulation system and platelet activation and aggregation and changing blood rheological conditions. Drugs used in anticoagulation therapy with anticoagulation, inhibiting platelet aggregation and associated with fibrinolysis, have been presented.
Natural products such as dietary supplements are important in ensuring health but have an important role in the prevention of cardiovascular and other diseases. Flavonoids as antioxidants reduce ROS levels and thus inhibit platelet aggregation/function or prothrombotic effects [125]. Flavonoids can alleviate ED, which is one of the factors leading to thrombosis, by inhibiting the excessive availability of TF in the endothelium [124]. Flavonoids can inhibit platelet activity bymple inhibiting the AA pathway [126] or may bind to the TXA2 platelet agonist and this way inhibit their aggregation.
Many flavonoids have anti-inflammatory, antioxidant, anti-cancer, and inhibitory properties related to platelet aggregation and adhesion. For example inhibition of platelet aggregation, were obtained using propolis, whose main ingredients are caffeic acid and its phenethyl ester, ferrulic acid, galangin, apigenin, quercetin, kaempferol, rutin, chrysin, pinostrobin, and pinocembrin [127].
In addition to flavonoids, isoflavonoids also show antiplatelet activity [128]. The use of wogonins and wogonoside led to inhibition of thrombin and FXa activity and inhibited thrombin production in human umbilical vein endothelial cells (HUVEC), as well as resulted in prolonged aPTT and PT. Furthermore, both flavonoids inhibited thrombin-catalysed fibrin polymerisation and platelet aggregation and showed anticoagulant activity in vivo in mice. Wogonin and wogonoside reduced the ratio of PAI-1 to t-PA. These properties testify to the good anticoagulant effect of both flavonoids [129]. For example, isoflavonoids genistein and daidzein, are COX-1 inhibitors and functional TXA2 receptor antagonists, affecting the platelet aggregation cascade [128]. Flavonolignans, silychristine, 1-benzofuran derivatives have antioxidant properties and are inhibitors of LOX, and prostaglandin synthetase [130]. They also inhibit platelet activation and decrease platelet aggregation and microparticle aggregation.
New natural substances with anticoagulant properties are currently being sought that would be free of side effects such as bleeding that may occur when using classic anticoagulants. Natural anticoagulants occur in the world of plants, which are traditionally used in Chinese medicine, but they also may be substances of animal origin [131]. The group of natural compounds with anticoagulant properties includes saponins, which occur in the world of plants and are produced by some marine organisms, including star fish and sea cucumbers. Saponins belong to amphipathic glycosides, which have one or more hydrophilic glycosides linked to a lipophilic triterpene or a steroid derivative [132]. Molecular weight depending on the basic backbone of the saponins and the number of sugar residues. Saponins have a high molecular weight ranging from 741 to 1808 g/mol [133].
For example Panax saponins inhibit platelet aggregation, preventing thrombosis, but also regulate many signalling pathways by acting on cardiovascular protection. In addition, they show anti-inflammatory effects mediated by peroxisome proliferator-activated receptor γ (PPAR-γ) in human endothelial cells. The main components of Panax notoginseng are ginsenoside Rg1, ginsenoside Rb1, and notoginsenoside R1. In vivo studied saponin Panax notoginseng reversed thrombin-induced hypercoagulable state saponins Panax induced PPAR-γ activation and also had a key role in the PI3Kβ/Akt/eNOS pathway in platelets [134]. Other PNS saponins have anti-inflammatory, antioxidant action, they inhibit platelet aggregation, regulate glucose and blood pressure, inhibit neuronal apoptosis, and provide neuronal protection [135].
Diosgenin and other steroidal saponins isolated from this plant are demonstrated to have anti-thrombotic activity. From few isolated saponins, especially Diosgenyl saponin, which has the linkage of the sugar chains at the C-3 position of diosgenin, had anti-thrombotic activity and significantly inhibited platelet aggregation in vitro and in vivo. This saponin did not act via the extrinsic coagulation pathway but rather by inhibiting molecular targets in intrinsic coagulation pathways [136].
Plant-derived diterpenoids including steviol derivatives demonstrate a wide range of biological activity and exhibit antitumor, antibacterial, antihyperglycemic, anti-inflammatory and immunomodulatory, antifungal, antiparasitic, antiviral, antiallergic, antispasmodic, antihyperglycemic, and other effects [137]. For example stevioside has been shown to inhibit the expression of IL-6, IL-1β and TNF-α and LPS-induced initiation, NF-κB, inhibitor of kappa B (IκBa) degradation, c-Jun N-terminal kinase (JNK), p38 and ERK phosphorylation in a dose-dependent manner. In addition, it was shown that the anti-inflammatory activity can be exerted by inhibiting NF-κB activation and MAPK and expression of pro-inflammatory cytokines [138]. Stevioside inhibited the release of proinflammatory cytokines and inhibited LPS-induced COX-2 and iNOS expression in mice in vivo [139].
8. Conclusions
This work reviews the pathophysiology of clot formation involving blood cells and cell components, critical factors released from activated cells and damaged cells as well as all already known drugs and methods to prevent or cure thromboembolism in all stages.
A new insight into the physiology of platelets and their involvement in inflammatory response and activation of clotting has been presented. These two processes have an important role in defending the host from infection. Their role in innate immunity in recognising pathogens, signal transduction, and cytokine/chemokine release is essential in thrombus formation, preventing the spread of microorganisms. In addition to platelets, other blood morphotic components such as leukocytes, erythrocytes, and vascular endothelial cells, along with activation of the coagulation system, have a significant role in the development of blood clots.
We considered the specific and key role of ECs in signalling mechanisms in which cyclophilin A (CypA) is secreted and ROS are generated. Both factors are sufficient to initiate vasculitis, secret MMPs, and induce changes in the structure of the vessel wall. The studies presented thus far show the role of platelets in the stages of inflammation, which indicates that they can be the target of an appropriate group of drugs. Undoubtedly, antiplatelet agents are effective in the treatment of thrombosis, but the problem is an increased risk of bleeding. However, a more accurate understanding of the processes associated with thrombosis and haemostasis has allowed the adoption of new strategies that are tested on animal models. Those are antiplatelet agents that inhibit thrombosis but do not affect haemostasis. In addition to antiplatelet drugs, there are also new strategies targeting clotting factors. An example is FXI, the inhibition of which may prove to be safer than the use of FXa inhibitors or thrombin. The FXI factor is important for the stabilisation and growth of the thrombus, but less important for haemostasis.
Even though modern medicine has a wide range of anticoagulant drugs, thrombosis still leads to cardiovascular diseases, myocardial infarction, and stroke. While significant advances have been made in understanding the mechanisms of thrombus formation and the pathophysiology of thrombosis, current prevention or treatment drugs that have been used for many years have been replaced by newer ones that show greater potential for the prevention and treatment of thrombosis with modest but gradual improvement. High hopes are associated with compounds of natural origin, which show a strong anticoagulant effect both at the experimental and clinical stage and can be used in the prophylactic or supportive methods of treatment of thrombotic diseases.
References
- Weitz, J.I.; Chan, N.C. Advances in Antithrombotic Therapy. Arteriosclerosis, Thrombosis, and Vascular Biology 2019, 39, 7–12.
- Essien, E.-O.; Rali, P.; Mathai, S.C. Pulmonary Embolism. Medical Clinics of North America 2019, 103, 549–564.
- Flumignan, C.D.; Flumignan, R.L.; Baptista-Silva, J.C. Antiplatelet agents for the treatment of deep venous thrombosis. Cochrane Database of Systematic Reviews 2016, 1–12.
- Wattanakit, K.; Lutsey, P.L.; Bell, E.J.; Gornik, H.; Cushman, M.; Heckbert, S.R.; Rosamond, W.D.; Folsom, A.R. Association between cardiovascular disease risk factors and occurrence of venous thromboembolism. Thrombosis and Haemostasis 2012, 108, 508–515.
- Stone, J.; Hangge, P.; Albadawi, H.; Wallace, A.; Shamoun, F.; Knuttien, M.G.; Naidu, S.; Oklu, R. Deep vein thrombosis: pathogenesis, diagnosis, and medical management. Cardiovascular Diagnosis and Therapy 2017, 7(Suppl 3), 276–284.
- Criqui, M.H.; Denenberg, J.O.; Bergan, J.; Langer, R.D.; Fronek, A. Risk factors for chronic venous disease: The San Diego Population Study. Journal of Vascular Surgery 2007, 46, 331–337.
- Kwiatkowska, W.; Knysz, B.; Gąsiorowski, J.; Witkiewicz, W. Deep vein thrombosis of the lower limbs in intravenous drug users. Postępy Higieny i Medycyny Doświadczalnej 2015, 69, 510–520.
- Pearson, J.D. 1 Endothelial cell function and thrombosis. Baillière’s Clinical Haematology 1994, 7, 441–452.
- van Hinsbergh, V.W.M. Endothelium—role in regulation of coagulation and inflammation. Seminars in Immunopathology 2012, 34, 93–106.
- Wolberg, A.S.; Campbell, R.A. Thrombin generation, fibrin clot formation and hemostasis. Transfusion and Apheresis Science 2008, 38, 15–23.
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell Biochem 2017, 82, 405–456.
- Petrillo, G.; Cirillo, P.; Luana D’Ascoli, G.-; Maresca, F.; Ziviello, F.; Chiariello, M. Tissue Factor/Factor FVII Complex Inhibitors in Cardiovascular Disease. Are Things Going Well? Current Cardiology Reviews 2010, 6, 325–332.
- Budnik, I.; Brill, A. Immune Factors in Deep Vein Thrombosis Initiation. Trends in Immunology 2018, 39, 610–623.
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: an update. European Heart Journal 2016, ehw550.
- Tanaka, K.A.; Key, N.S.; Levy, J.H. Blood Coagulation: Hemostasis and Thrombin Regulation. Anesthesia & Analgesia 2009, 108, 1433–1446.
- MCKenzie, P.J. Deep venous thrombosis and anaesthesia. British Journal of Anaesthesia 1991, 67, 128.
- Branchford, B.R.; Carpenter, S.L. The Role of Inflammation in Venous Thromboembolism. Frontiers in Pediatrics 2018, 6, 1–7.
- Iba, T.; Levy, J.H. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. Journal of Thrombosis and Haemostasis 2018, 16, 231–241.
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Scientific Reports 2019, 9, 13397.
- Waldron, B.; Moll, S. A patient’s guide to recovery after deep vein thrombosis or pulmonary embolism. Circulation 2014, 129, 477–479.
- Sen, C.K.; Roy, S. Wound healing. Plastic surgery 2012.
- Korzonek-Szlacheta, I.; Zubelewicz-Szkodzińska, B.; Gąsior, M. Płytki krwi — ogniwo łączące zakrzepicę ze stanem zapalnym. Folia Cardiologica 2018, 13, 303–308.
- Violi, F.; Pignatelli, P. Platelet Oxidative Stress and Thrombosis. Thrombosis Research 2012, 129, 378–381.
- Seno, T.; Inoue, N.; Gao, D.; Okuda, M.; Sumi, Y.; Matsui, K.; Yamada, S.; Hirata, K.; Kawashima, S.; Tawa, R.; et al. Involvement of NADH/NADPH Oxidase in Human Platelet ROS Production. Thrombosis Research 2001, 103, 399–409.
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biology 2018, 14, 126–130.
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Thomas, M.R.; Henderson, I.R.; Watson, S.P.; Cunningham, A.F. Understanding Infection-Induced Thrombosis: Lessons Learned From Animal Models. Frontiers in Immunology 2019, 10, 17.
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2000 2010, 54, 15–44.
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Frontiers in Cardiovascular Medicine 2019, 6, 132.
- Montoro-García, S.; Schindewolf, M.; Stanford, S.; Larsen, O.; Thiele, T. The Role of Platelets in Venous Thromboembolism. Seminars in Thrombosis and Hemostasis 2016, 42, 242–251.
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. Journal of the American College of Cardiology 2003, 42, 1149–1160.
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. Journal of Vascular Surgery: Venous and Lymphatic Disorders 2018, 6, 552–553.
- Echeverría, C.; Montorfano, I.; Hermosilla, T.; Armisén, R.; Velásquez, L.A.; Cabello-Verrugio, C.; Varela, D.; Simon, F. Endotoxin Induces Fibrosis in Vascular Endothelial Cells through a Mechanism Dependent on Transient Receptor Protein Melastatin 7 Activity. PLoS ONE 2014, 9, e94146.
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and Cytokine Receptors in Advanced Heart Failure. Circulation 2001, 103, 2055–2059.
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascular Pharmacology 2018.
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–61.
- Fioranelli, M.; Bottaccioli, A.G.; Bottaccioli, F.; Bianchi, M.; Rovesti, M.; Roccia, M.G. Stress and Inflammation in Coronary Artery Disease: A Review Psychoneuroendocrineimmunology-Based. Frontiers in Immunology 2018, 9, 2031.
- Allen, R.C.; Stephens, J.T. Myeloperoxidase Selectively Binds and Selectively Kills Microbes. Infection and Immunity 2011, 79, 474–485.
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS Journal 2008, 275, 3278–3289.
- Spooner, R.; Yilmaz, Ö. The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation. International Journal of Molecular Sciences 2011, 12, 334–352.
- Case, A. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, 82.
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Biology and Medicine 2016, 100, 14–31.
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian Journal of Clinical Biochemistry 2015, 30, 11–26.
- Bielski, B.H.J.; Cabelli, D.E. Superoxide and Hydroxyl Radical Chemistry in Aqueous Solution. In Active Oxygen in Chemistry; Springer Netherlands: Dordrecht, 1995; pp. 66–104.
- Li, Y.; Zhu, H.; Kuppusamy, P.; Zweier, J.; Trush, M. Mitochondrial Electron Transport Chain-Derived Superoxide Exits Macrophages: Implications for Mononuclear Cell-Mediated Pathophysiological Processes. Reactive Oxygen Species 2016, 1, 81–98.
- Babior, B.M. NADPH oxidase. Current Opinion in Immunology 2004, 16, 42–47.
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochimica et Biophysica Acta (BBA) - Biomembranes 2006, 1758, 994–1003.
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. Journal of Leukocyte Biology 2004, 76, 760–781.
- Barrett, C.D.; Hsu, A.T.; Ellson, C.D.; Y.Miyazawa, B.; Kong, Y.-W.; Greenwood, J.D.; Dhara, S.; Neal, M.D.; Sperry, J.L.; Park, M.S.; et al. Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clinical & Experimental Immunology 2018, 194, 103–117.
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 2017, 37, 41–52.
- Galley, J.C.; Straub, A.C. Redox Control of Vascular Function. Arteriosclerosis, Thrombosis, and Vascular Biology 2017, 37, 178–184.
- Aldosari, S.; Awad, M.; Harrington, E.; Sellke, F.; Abid, M. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14.
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. British Journal of Dermatology 2010, 163, 257–268.
- Arvieux, J.; Regnault, V.; Hachulla, E.; Darnige, L.; Berthou, F.; Youinou, P. Oxidation of beta2-glycoprotein I (beta2GPI) by the hydroxyl radical alters phospholipid binding and modulates recognition by anti-beta2GPI autoantibodies. Arthritis Research & Therapy 2001, 3, P082.
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. Journal of Physiology 2003, 552, 335–344.
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radical Biology and Medicine 2007, 43, 332–347.
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arteriosclerosis, Thrombosis, and Vascular Biology 2010, 30, 653–661.
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circulation Research 2008, 102, 488–496.
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J.R. From form to function: the role of Nox4 in the cardiovascular system. Frontiers in Physiology 2012, 3, 412.
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. Journal of Clinical Medicine 2017, 6, 22.
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutation Research/Reviews in Mutation Research 2012, 751, 77–81.
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. American Journal of Physiology-Cell Physiology 2017, 312, 749–764.
- Saini, R.; Singh, S. Inducible nitric oxide synthase: An asset to neutrophils. Journal of Leukocyte Biology 2019, 105, 49–61.
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proceedings of the National Academy of Sciences 2010, 107, 15880–15885.
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Letters 2010, 584, 3193–3197.
- Dikshit, M.; Kumari, R.; Srimal, R.C. Pulmonary thromboembolism-induced alterations in nitric oxide release from rat circulating neutrophils. The Journal of pharmacology and experimental therapeutics 1993, 265, 1369–73.
- Mitsuke, Y.; Lee, J.-D.; Shimizu, H.; Uzui, H.; Iwasaki, H.; Ueda, T. Nitric oxide synthase activity in peripheral polymorphonuclear leukocytes in patients with chronic congestive heart failure. The American Journal of Cardiology 2001, 87, 183–187.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Frontiers in Cellular and Infection Microbiology 2017, 7, 1–24.
- Kirchner, T.; Möller, S.; Klinger, M.; Solbach, W.; Laskay, T.; Behnen, M. The Impact of Various Reactive Oxygen Species on the Formation of Neutrophil Extracellular Traps. Mediators of Inflammation 2012, 2012, 1–10.
- Paiva, C.N.; Bozza, M.T. Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxidants & Redox Signaling 2014, 20, 1000–1037.
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.-C.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunological Reviews 2016, 273, 180–193.
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circulation Research 2001, 88, 888–894.
- San José, G.; Bidegain, J.; Robador, P.A.; Díez, J.; Fortuño, A.; Zalba, G. Insulin-induced NADPH oxidase activation promotes proliferation and matrix metalloproteinase activation in monocytes/macrophages. Free Radical Biology and Medicine 2009, 46, 1058–1067.
- Xue, C.; Sowden, M.P.; Berk, B.C. Extracellular and Intracellular Cyclophilin A, Native and Post-Translationally Modified, Show Diverse and Specific Pathological Roles in Diseases. Arteriosclerosis, Thrombosis, and Vascular Biology 2018, 38, 986–993.
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Research in Cardiology 2008, 103, 398–406.
- Yu, W.; Hu, X.; Wang, M. Pterostilbene inhibited advanced glycation end products (AGEs)-induced oxidative stress and inflammation by regulation of RAGE/MAPK/NF-κB in RAW264.7 cells. Journal of Functional Foods 2018, 40, 272–279.
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Frontiers in Immunology 2014, 5, 1–14.
- Alvarez, B.; Radi, R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids 2003, 25, 295–311.
- Elsayed, N.M. Toxicity of nitrogen dioxide: an introduction. Toxicology 1994, 89, 161–174.
- Persinger, R.L.; Poynter, M.E.; Ckless, K.; Janssen-Heininger, Y.M.W. Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung. Molecular and Cellular Biochemistry 2002, 234, 71–80.
- Gray, B.; Carmichael, A.J. Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance. Biochemical Journal 1992, 281, 795–802.
- Crow, J.P.; Beckman, J.S. Reactions between Nitric Oxide, Superoxide, and Peroxynitrite: Footprints of Peroxynitrite in Vivo. Advances in Pharmacology 1995, 34, 17–43.
- Nauser, T.; Koppenol, W.H. The Rate Constant of the Reaction of Superoxide with Nitrogen Monoxide: Approaching the Diffusion Limit. The Journal of Physical Chemistry A 2002, 106, 4084–4086.
- Münzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Annals of Medicine 2008, 40, 180–196.
- Gow, A.J.; Luchsinger, B.P.; Pawloski, J.R.; Singel, D.J.; Stamler, J.S. The oxyhemoglobin reaction of nitric oxide. Proceedings of the National Academy of Sciences 1999, 96, 9027–9032.
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatric Research 2009, 66, 121–127.
- BRZESZCZYNSKA, J.; GWOZDZINSKI, K. Nitric oxide induced oxidative changes in erythrocyte membrane components. Cell Biology International 2008, 32, 114–120.
- Ansari, S.A.; Pendurthi, U.R.; Rao, L.V.M. The lipid peroxidation product 4-hydroxy-2-nonenal induces tissue factor decryption via ROS generation and the thioredoxin system. Blood Advances 2017, 1, 2399–2413.
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive Oxygen Species in Venous Thrombosis. International Journal of Molecular Sciences 2020, 21, 1918.
- Golino, P.; Ragni, M.; Cirillo, P.; Avvdimento, V.E.; Feliciello, A.; Esposito, N.; Scognamiglio, A.; Trimarco, B.; Iaccarino, G.; Condorelli, M.; et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nature Medicine 1996, 2, 35–40.
- Cadroy, Y.; Dupouy, D.; Boneu, B.; Plaisancié, H. Polymorphonuclear Leukocytes Modulate Tissue Factor Production by Mononuclear Cells: Role of Reactive Oxygen Species. The Journal of Immunology 2000, 164, 3822–3828.
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Görlach, A. NADPH Oxidase Mediates Tissue Factor–Dependent Surface Procoagulant Activity by Thrombin in Human Vascular Smooth Muscle Cells. Circulation 2002, 105, 2030–2036.
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918.
- Lichota, A.; Gwozdzinski, L.; Gwozdzinski, K. Therapeutic potential of natural compounds in inflammation and chronic venous insufficiency. European Journal of Medicinal Chemistry 2019, 176, 68–91.
- Kovacs, M.J.; Kahn, S.R.; Wells, P.S.; Anderson, D.A.; Chagnon, I.; Le Gal, G.; Solymoss, S.; Crowther, M.; Perrier, A.; Ramsay, T.; et al. Patients with a first symptomatic unprovoked deep vein thrombosis are at higher risk of recurrent venous thromboembolism than patients with a first unprovoked pulmonary embolism. Journal of Thrombosis and Haemostasis 2010, 8, 1926–1932.
- Ringleb, P.A. Thrombolytics, Anticoagulants, and Antiplatelet Agents. Stroke 2006, 37, 312–313.
- Lee, W.; Lee, H.; Kim, M.-A.; Choi, J.; Kim, K.-M.; Hwang, J.S.; Na, M.; Bae, J.-S. Evaluation of novel factor Xa inhibitors from Oxya chinensis sinuosa with anti-platelet aggregation activity. Scientific Reports 2017, 7, 7934.
- Fischer, P.M. Design of Small-Molecule Active-Site Inhibitors of the S1A Family Proteases as Procoagulant and Anticoagulant Drugs. Journal of Medicinal Chemistry 2018, 61, 3799–3822.
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic Review. Clinical Pharmacokinetics 2019, 58, 1265–1279.
- Bratsos, S. Pharmacokinetic Properties of Rivaroxaban in Healthy Human Subjects. Cureus 2019, 11, e5484.
- Rao, P.S.S.; Burkart, T. Advances in oral anticoagulation therapy – What’s in the pipeline? Blood Reviews 2017, 31, 205–211.
- Kamikubo, Y.; Mendolicchio, G.L.; Zampolli, A.; Marchese, P.; Rothmeier, A.S.; Orje, J.N.; Gale, A.J.; Krishnaswamy, S.; Gruber, A.; Østergaard, H.; et al. Selective factor VIII activation by the tissue factor–factor VIIa–factor Xa complex. Blood 2017, 130, 1661–1670.
- Capodanno, D.; Ferreiro, J.L.; Angiolillo, D.J. Antiplatelet therapy: new pharmacological agents and changing paradigms. Journal of Thrombosis and Haemostasis 2013, 11, 316–329.
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: emphasis on preserving haemostasis. Nature Reviews Cardiology 2018, 15, 181–191.
- Harter, K.; Levine, M.; Henderson, S. Anticoagulation Drug Therapy: A Review. Western Journal of Emergency Medicine 2015, 16, 11–17.
- Rubboli, A.; Patti, G. What is the Role for Glycoprotein IIB/IIIA Inhibitor Use in the Catheterization Laboratory in the Current Era? Current Vascular Pharmacology 2018, 16, 451–458.
- Quinn, M.J.; Fitzgerald, D.J. Ticlopidine and Clopidogrel. Circulation 1999, 100, 1667–1672.
- Jiang, M.; You, J.H. Review of pharmacoeconomic evaluation of genotype-guided antiplatelet therapy. Expert Opinion on Pharmacotherapy 2015, 16, 771–779.
- Storey, R.F.; Gurbel, P.A.; ten Berg, J.; Bernaud, C.; Dangas, G.D.; Frenoux, J.-M.; Gorog, D.A.; Hmissi, A.; Kunadian, V.; James, S.K.; et al. Pharmacodynamics, pharmacokinetics, and safety of single-dose subcutaneous administration of selatogrel, a novel P2Y12 receptor antagonist, in patients with chronic coronary syndromes. European Heart Journal 2020, 41, 3132–3140.
- Sturgeon, S.A.; Jones, C.; Angus, J.A.; Wright, C.E. Advantages of a selective β-isoform phosphoinositide 3-kinase antagonist, an anti-thrombotic agent devoid of other cardiovascular actions in the rat. European Journal of Pharmacology 2008, 587, 209–215.
- Blair, T.A.; Moore, S.F.; Williams, C.M.; Poole, A.W.; Vanhaesebroeck, B.; Hers, I. Phosphoinositide 3-Kinases p110α and p110β Have Differential Roles in Insulin-Like Growth Factor-1–Mediated Akt Phosphorylation and Platelet Priming. Arteriosclerosis, Thrombosis, and Vascular Biology 2014, 34, 1681–1688.
- Laurent, P.-A.; Séverin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.-P. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888.
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clinical and Translational Medicine 2020, 9, 8.
- Gilbert, J.C.; DeFeo-Fraulini, T.; Hutabarat, R.M.; Horvath, C.J.; Merlino, P.G.; Marsh, H.N.; Healy, J.M.; BouFakhreddine, S.; Holohan, T. V.; Schaub, R.G. First-in-Human Evaluation of Anti–von Willebrand Factor Therapeutic Aptamer ARC1779 in Healthy Volunteers. Circulation 2007, 116, 2678–2686.
- Huang, R.-H.; Fremont, D.H.; Diener, J.L.; Schaub, R.G.; Sadler, J.E. A Structural Explanation for the Antithrombotic Activity of ARC1172, a DNA Aptamer that Binds von Willebrand Factor Domain A1. Structure 2009, 17, 1476–1484.
- Hashemzadeh, M.; Furukawa, M.; Goldsberry, S.; Movahed, M.R. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Experimental and clinical cardiology 2008, 13, 192–7.
- Hanlon, A.; Metjian, A. Caplacizumab in adult patients with acquired thrombotic thrombocytopenic purpura. Therapeutic Advances in Hematology 2020, 11, 204062072090290.
- Bartunek, J.; Barbato, E.; Heyndrickx, G.; Vanderheyden, M.; Wijns, W.; Holz, J.-B. Novel Antiplatelet Agents: ALX-0081, a Nanobody Directed towards von Willebrand Factor. Journal of Cardiovascular Translational Research 2013, 6, 355–363.
- Zheng, L.; Mao, Y.; Abdelgawwad, M.S.; Kocher, N.K.; Li, M.; Dai, X.; Li, B.; Zheng, X.L. Therapeutic efficacy of the platelet glycoprotein Ib antagonist anfibatide in murine models of thrombotic thrombocytopenic purpura. Blood Advances 2016, 1, 75–83.
- Kong, Y.; Huo, J.; Xu, W.; Xiong, J.; Li, Y.; Wu, W. A novel anti-platelet aggregation tripeptide from Agkistrodon acutus venom: Isolation and characterization. Toxicon 2009, 54, 103–109.
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thrombosis and Haemostasis 2014, 111, 279–289.
- Li, T.-T.; Fan, M.-L.; Hou, S.-X.; Li, X.-Y.; Barry, D.M.; Jin, H.; Luo, S.-Y.; Kong, F.; Lau, L.-F.; Dai, X.-R.; et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. British Journal of Pharmacology 2015, 172, 3904–3916.
- Gailani, D.; Gruber, A. Factor XI as a Therapeutic Target. Arteriosclerosis, Thrombosis, and Vascular Biology 2016, 36, 1316–1322.
- Phang, M.; Lazarus, S.; Wood, L.; Garg, M. Diet and Thrombosis Risk: Nutrients for Prevention of Thrombotic Disease. Seminars in Thrombosis and Hemostasis 2011, 37, 199–208.
- Vazhappilly, C.G.; Ansari, S.A.; Al-Jaleeli, R.; Al-Azawi, A.M.; Ramadan, W.S.; Menon, V.; Hodeify, R.; Siddiqui, S.S.; Merheb, M.; Matar, R.; et al. Role of flavonoids in thrombotic, cardiovascular, and inflammatory diseases. Inflammopharmacology 2019, 27, 863–869.
- Freedman, J.E. Oxidative Stress and Platelets. Arteriosclerosis, Thrombosis, and Vascular Biology 2008, 28, 11–16.
- Faggio, C.; Sureda, A.; Morabito, S.; Sanches-Silva, A.; Mocan, A.; Nabavi, S.F.; Nabavi, S.M. Flavonoids and platelet aggregation: A brief review. European Journal of Pharmacology 2017, 807, 91–101.
- Zhang, Y.-X.; Yang, T.-T.; Xia, L.; Zhang, W.-F.; Wang, J.-F.; Wu, Y.-P. Inhibitory Effect of Propolis on Platelet Aggregation In Vitro. Journal of Healthcare Engineering 2017, 2017, 1–6.
- Karlíčková, J.; Říha, M.; Filipský, T.; Macáková, K.; Hrdina, R.; Mladěnka, P. Antiplatelet Effects of Flavonoids Mediated by Inhibition of Arachidonic Acid Based Pathway. Planta Medica 2015, 82, 76–83.
- Ku, S.-K.; Bae, J.-S. Antithrombotic activities of wogonin and wogonoside via inhibiting platelet aggregation. Fitoterapia 2014, 98, 27–35.
- Dasgupta, A. Antiinflammatory Herbal Supplements. In Translational Inflammation; Elsevier, 2019; pp. 69–91.
- Chen, C.; Yang, F.-Q.; Zhang, Q.; Wang, F.-Q.; Hu, Y.-J.; Xia, Z.-N. Natural Products for Antithrombosis. Evidence-Based Complementary and Alternative Medicine 2015, 2015, 1–17.
- Kregiel, D.; Berlowska, J.; Witonska, I.; Antolak, H.; Proestos, C.; Babic, M.; Babic, L.; Zhang, B. Saponin-Based, Biological-Active Surfactants from Plants. In Application and Characterization of Surfactants; InTech, 2017.
- Singh, D.; Chaudhuri, P.K. Structural characteristics, bioavailability and cardioprotective potential of saponins. Integrative Medicine Research 2018, 7, 33–43.
- Shen, Q.; Li, J.; Zhang, C.; Wang, P.; Mohammed, A.; Ni, S.; Tang, Z. Panax notoginseng saponins reduce high-risk factors for thrombosis through peroxisome proliferator-activated receptor -γ pathway. Biomedicine & Pharmacotherapy 2017, 96, 1163–1169.
- Xie, W.; Meng, X.; Zhai, Y.; Zhou, P.; Ye, T.; Wang, Z.; Sun, G.; Sun, X. Panax Notoginseng Saponins: A Review of Its Mechanisms of Antidepressant or Anxiolytic Effects and Network Analysis on Phytochemistry and Pharmacology. Molecules 2018, 23, 940.
- Zhang, R.; Huang, B.; Du, D.; Guo, X.; Xin, G.; Xing, Z.; Liang, Y.; Chen, Y.; Chen, Q.; He, Y.; et al. Anti-thrombosis effect of diosgenyl saponins in vitro and in vivo. Steroids 2013, 78, 1064–1070.
- Wang, M.; Li, H.; Xu, F.; Gao, X.; Li, J.; Xu, S.; Zhang, D.; Wu, X.; Xu, J.; Hua, H.; et al. Diterpenoid lead stevioside and its hydrolysis products steviol and isosteviol: Biological activity and structural modification. European Journal of Medicinal Chemistry 2018, 156, 885–906.
- Boonkaewwan, C.; Burodom, A. Anti-inflammatory and immunomodulatory activities of stevioside and steviol on colonic epithelial cells. Journal of the Science of Food and Agriculture 2013, 93, 3820–3825.
- Yingkun, N.; Zhenyu, W.; Jing, L.; Xiuyun, L.; Huimin, Y. Stevioside Protects LPS-Induced Acute Lung Injury in Mice. Inflammation 2013, 36, 242–250.

