 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Estelle A. Duprez | -- | 3139 | 2022-10-17 18:06:03 | | | |
| 2 | Jessie Wu | + 9 word(s) | 3148 | 2022-10-18 07:19:27 | | | | |
| 3 | Jessie Wu | Meta information modification | 3148 | 2022-10-18 07:21:42 | | | | |
| 4 | Jessie Wu | Meta information modification | 3148 | 2022-10-18 07:22:39 | | |
Video Upload Options
Aging leads to a decline in the functions of the hematopoietic and immune system, which in the elderly results in an increased risk of infection, poor vaccination efficacy, anemia and blood cancers. It is now well established that age-related dysfunction of the entire hematopoietic system originates from hematopoietic stem cells (HSCs), which lose their fitness over time. Single-cell transcriptomic technologies enable the uncovering and characterization of cellular heterogeneity and pave the way for studies aiming at understanding the origin and consequences of it. The hematopoietic system is in essence a very well adapted model system to benefit from this technological advance because it is characterized by different cellular states. Each cellular state, and its interconnection, may be defined by a specific location in the global transcriptional landscape sustained by a complex regulatory network. This transcriptomic signature is not fixed and evolved over time to give rise to less efficient hematopoietic stem cells (HSC), leading to a well-documented hematopoietic aging.
1. Understanding Hematopoietic Stem Cell Heterogeneity to Grasp Aged Hematopoietic Stem Cell Deregulations
2. Coupling the Transcriptome to the Cell Fate
3. Network-Based Dynamic Modeling: A Successful Approach to Decipher Hematopoiesis
4. Single Cell Data and Boolean Networks Inference to Understand Hematopoietic Stem Cell Aging
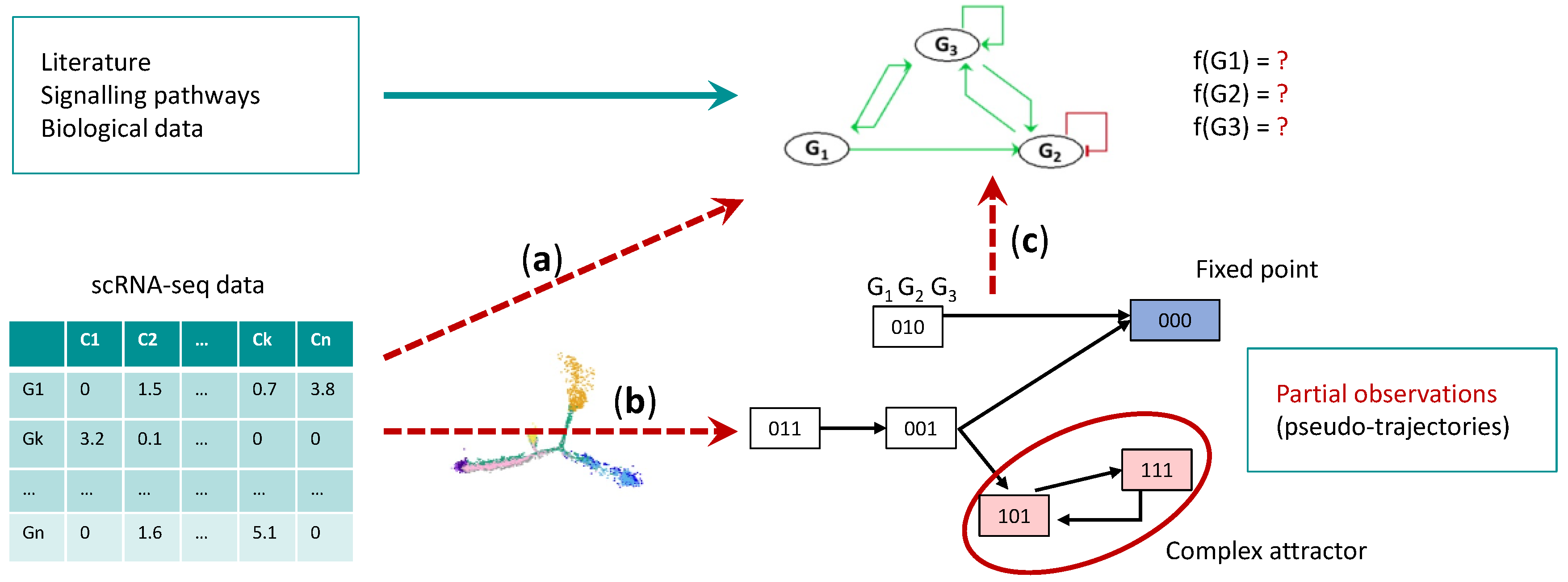
4.1. Gene Regulatory Network Inference
4.2. Boolean Network Inference
References
- Haas, S.; Trumpp, A.; Milsom, M.D. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 2018, 22, 627–638.
- Zhang, Y.; Gao, S.; Xia, J.; Liu, F. Hematopoietic Hierarchy—An Updated Roadmap. Trends Cell Biol. 2018, 28, 976–986.
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426.
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottgens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31.
- Herman, J.S.; Sagar; Grun, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat. Methods 2018, 15, 379–386.
- Herault, L.; Poplineau, M.; Mazuel, A.; Platet, N.; Remy, E.; Duprez, E. Single-cell RNA-seq reveals a concomitant delay in differentiation and cell cycle of aged hematopoietic stem cells. BMC Biol. 2021, 19, 19.
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216.
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116.
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281.
- Waddington, C.H.; Kacser, H. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology; Allen & Unwin: Crows Nest, Australia, 1957; pp. 42–46.
- Buenrostro, J.D.; Corces, M.R.; Lareau, C.A.; Wu, B.; Schep, A.N.; Aryee, M.J.; Majeti, R.; Chang, H.Y.; Greenleaf, W.J. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 2018, 173, 1535–1548.e16.
- Young, K.; Borikar, S.; Bell, R.; Kuffler, L.; Philip, V.; Trowbridge, J.J. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J. Exp. Med. 2016, 213, 2259–2267.
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075.
- Mann, M.; Mehta, A.; de Boer, C.G.; Kowalczyk, M.S.; Lee, K.; Haldeman, P.; Rogel, N.; Knecht, A.R.; Farouq, D.; Regev, A.; et al. Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep. 2018, 25, 2992–3005.e5.
- Fast, E.M.; Sporrij, A.; Manning, M.; Rocha, E.L.; Yang, S.; Zhou, Y.; Guo, J.; Baryawno, N.; Barkas, N.; Scadden, D.; et al. External signals regulate continuous transcriptional states in hematopoietic stem cells. eLife 2021, 10, e66512.
- Sommerkamp, P.; Romero-Mulero, M.C.; Narr, A.; Ladel, L.; Hustin, L.; Schonberger, K.; Renders, S.; Altamura, S.; Zeisberger, P.; Jacklein, K.; et al. Mouse multipotent progenitor 5 cells are located at the interphase between hematopoietic stem and progenitor cells. Blood 2021, 137, 3218–3224.
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874.
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 2010, 6, 265–278.
- Qiu, J.; Papatsenko, D.; Niu, X.; Schaniel, C.; Moore, K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. 2014, 2, 473–490.
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015, 25, 1860–1872.
- Kirschner, K.; Chandra, T.; Kiselev, V.; Flores-Santa Cruz, D.; Macaulay, I.C.; Park, H.J.; Li, J.; Kent, D.G.; Kumar, R.; Pask, D.C.; et al. Proliferation Drives Aging-Related Functional Decline in a Subpopulation of the Hematopoietic Stem Cell Compartment. Cell Rep. 2017, 19, 1503–1511.
- Desterke, C.; Bennaceur-Griscelli, A.; Turhan, A.G. EGR1 dysregulation defines an inflammatory and leukemic program in cell trajectory of human-aged hematopoietic stem cells (HSC). Stem Cell Res. Ther. 2021, 12, 419.
- Passegue, E.; Wagner, E.F.; Weissman, I.L. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 2004, 119, 431–443.
- Santaguida, M.; Schepers, K.; King, B.; Sabnis, A.J.; Forsberg, E.C.; Attema, J.L.; Braun, B.S.; Passegue, E. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 2009, 15, 341–352.
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e19.
- Lauridsen, F.K.B.; Jensen, T.L.; Rapin, N.; Aslan, D.; Wilhelmson, A.S.; Pundhir, S.; Rehn, M.; Paul, F.; Giladi, A.; Hasemann, M.S.; et al. Differences in Cell Cycle Status Underlie Transcriptional Heterogeneity in the HSC Compartment. Cell Rep. 2018, 24, 766–780.
- Florian, M.C.; Klose, M.; Sacma, M.; Jablanovic, J.; Knudson, L.; Nattamai, K.J.; Marka, G.; Vollmer, A.; Soller, K.; Sakk, V.; et al. Aging alters the epigenetic asymmetry of HSC division. PLoS Biol. 2018, 16, e2003389.
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160.
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673.
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. eLife 2017, 6, e27041.
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021, 374, abe6474.
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lonnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498.
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020, 38, 1408–1414.
- Bowman, T.V.; McCooey, A.J.; Merchant, A.A.; Ramos, C.A.; Fonseca, P.; Poindexter, A.; Bradfute, S.B.; Oliveira, D.M.; Green, R.; Zheng, Y.; et al. Differential mRNA processing in hematopoietic stem cells. Stem Cells 2006, 24, 662–670.
- Bergen, V.; Soldatov, R.A.; Kharchenko, P.V.; Theis, F.J. RNA velocity-current challenges and future perspectives. Mol. Syst. Biol. 2021, 17, e10282.
- Tak, T.; Eisele, A.S.; Perie, L. In Vivo Tracking of Hematopoietic Stem and Progenitor Cell Ontogeny by Cellular Barcoding. Methods Mol. Biol. 2021, 2308, 281–300.
- Rodriguez-Fraticelli, A.E.; Camargo, F. Systems analysis of hematopoiesis using single-cell lineage tracing. Curr. Opin. Hematol. 2021, 28, 18–27.
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381.
- Rodriguez-Fraticelli, A.E.; Weinreb, C.; Wang, S.W.; Migueles, R.P.; Jankovic, M.; Usart, M.; Klein, A.M.; Lowell, S.; Camargo, F.D. Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature 2020, 583, 585–589.
- Yu, V.W.C.; Yusuf, R.Z.; Oki, T.; Wu, J.; Saez, B.; Wang, X.; Cook, C.; Baryawno, N.; Ziller, M.J.; Lee, E.; et al. Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 2016, 167, 1310–1322.e17.
- Whichard, Z.L.; Sarkar, C.A.; Kimmel, M.; Corey, S.J. Hematopoiesis and its disorders: A systems biology approach. Blood 2010, 115, 2339–2347.
- Wichmann, H.E.; Loeffler, M.; Schmitz, S. A concept of hemopoietic regulation and its biomathematical realization. Blood Cells 1988, 14, 411–429.
- Mackey, M.C. Mathematical Models of Hematopoietic Cell Replication and Control; Prentice-Hall, Inc.: Hoboken, NJ, USA, 1997.
- Manesso, E.; Teles, J.; Bryder, D.; Peterson, C. Dynamical modelling of haematopoiesis: An integrated view over the system in homeostasis and under perturbation. J. R. Soc. Interface 2013, 10, 20120817.
- Le Novere, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158.
- Calzone, L.; Barillot, E.; Zinovyev, A. Logical versus kinetic modeling of biological networks: Applications in cancer research. Curr. Opin. Chem. Eng. 2018, 21, 22–31.
- Albert, R.; Thakar, J. Boolean modeling: A logic-based dynamic approach for understanding signaling and regulatory networks and for making useful predictions. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 353–369.
- Choi, M.; Shi, J.; Jung, S.H.; Chen, X.; Cho, K.H. Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci. Signal. 2012, 5, ra83.
- Remy, E.; Rebouissou, S.; Chaouiya, C.; Zinovyev, A.; Radvanyi, F.; Calzone, L. A Modeling Approach to Explain Mutually Exclusive and Co-Occurring Genetic Alterations in Bladder Tumorigenesis. Cancer Res. 2015, 75, 4042–4052.
- Naldi, A.; Carneiro, J.; Chaouiya, C.; Thieffry, D. Diversity and plasticity of Th cell types predicted from regulatory network modelling. PLoS Comput. Biol. 2010, 6, e1000912.
- Cacace, E.; Collombet, S.; Thieffry, D. Logical modeling of cell fate specification-Application to T cell commitment. Curr. Top. Dev. Biol. 2020, 139, 205–238.
- Rodriguez-Jorge, O.; Kempis-Calanis, L.A.; Abou-Jaoude, W.; Gutierrez-Reyna, D.Y.; Hernandez, C.; Ramirez-Pliego, O.; Thomas-Chollier, M.; Spicuglia, S.; Santana, M.A.; Thieffry, D. Cooperation between T cell receptor and Toll-like receptor 5 signaling for CD4(+) T cell activation. Sci. Signal. 2019, 12, eaar3641.
- Kondratova, M.; Barillot, E.; Zinovyev, A.; Calzone, L. Modelling of Immune Checkpoint Network Explains Synergistic Effects of Combined Immune Checkpoint Inhibitor Therapy and the Impact of Cytokines in Patient Response. Cancers 2020, 12, 3600.
- Krumsiek, J.; Marr, C.; Schroeder, T.; Theis, F.J. Hierarchical differentiation of myeloid progenitors is encoded in the transcription factor network. PLoS ONE 2011, 6, e22649.
- Bonzanni, N.; Garg, A.; Feenstra, K.A.; Schutte, J.; Kinston, S.; Miranda-Saavedra, D.; Heringa, J.; Xenarios, I.; Gottgens, B. Hard-wired heterogeneity in blood stem cells revealed using a dynamic regulatory network model. Bioinformatics 2013, 29, i80–i88.
- Collombet, S.; van Oevelen, C.; Ortega, J.L.S.; Abou-Jaoude, W.; Di Stefano, B.; Thomas-Chollier, M.; Graf, T.; Thieffry, D. Logical modeling of lymphoid and myeloid cell specification and transdifferentiation. Proc. Natl. Acad. Sci. USA 2017, 114, 5792–5799.
- Enciso, J.; Mayani, H.; Mendoza, L.; Pelayo, R. Modeling the Pro-inflammatory Tumor Microenvironment in Acute Lymphoblastic Leukemia Predicts a Breakdown of Hematopoietic-Mesenchymal Communication Networks. Front. Physiol. 2016, 7, 349.
- Ikonomi, N.; Kuhlwein, S.D.; Schwab, J.D.; Kestler, H.A. Awakening the HSC: Dynamic Modeling of HSC Maintenance Unravels Regulation of the TP53 Pathway and Quiescence. Front. Physiol. 2020, 11, 848.
- Marbach, D.; Roy, S.; Ay, F.; Meyer, P.E.; Candeias, R.; Kahveci, T.; Bristow, C.A.; Kellis, M. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 2012, 22, 1334–1349.
- Hu, X.; Hu, Y.; Wu, F.; Leung, R.W.T.; Qin, J. Integration of single-cell multi-omics for gene regulatory network inference. Comput. Struct. Biotechnol. J. 2020, 18, 1925–1938.
- Pratapa, A.; Jalihal, A.P.; Law, J.N.; Bharadwaj, A.; Murali, T.M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 2020, 17, 147–154.
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776.
- Gao, N.P.; Ud-Dean, S.M.M.; Gandrillon, O.; Gunawan, R. SINCERITIES: Inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 2018, 34, 258–266.
- Specht, A.T.; Li, J. LEAP: Constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics 2017, 33, 764–766.
- Chan, T.E.; Stumpf, M.P.H.; Babtie, A.C. Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst. 2017, 5, 251–267.e3.
- Verny, L.; Sella, N.; Affeldt, S.; Singh, P.P.; Isambert, H. Learning causal networks with latent variables from multivariate information in genomic data. PLoS Comput. Biol. 2017, 13, e1005662.
- Herrmann, F.; Gross, A.; Zhou, D.; Kestler, H.A.; Kuhl, M. A boolean model of the cardiac gene regulatory network determining first and second heart field identity. PLoS ONE 2012, 7, e46798.
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086.
- Arguelaguet, R.; Lohoff, T.; Li, J.G.; Nakhuda, A.; Krueger, F.; Velten, L.; Clarck, S.J.; Reik, W. Decoding gene regulation in the mouse embryo using single-cell multi-omics. bioRxiv 2022.
- Moignard, V.; Woodhouse, S.; Haghverdi, L.; Lilly, A.J.; Tanaka, Y.; Wilkinson, A.C.; Buettner, F.; Macaulay, I.C.; Jawaid, W.; Diamanti, E.; et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol. 2015, 33, 269–276.
- Chevalier, S.; Froidevaux, C.; Paulevé, L.; Zinovyev, A. Synthesis of Boolean networks from biological dynamical constraints using answer-set programming. In Proceedings of the IEEE 31st Internationnal Conference on Tools with Artificial Intelligence, Portland, OR, USA, 4–6 November 2019; pp. 34–41.
- Ostrowski, M.; Pauleve, L.; Schaub, T.; Siegel, A.; Guziolowski, C. Boolean network identification from perturbation time series data combining dynamics abstraction and logic programming. Biosystems 2016, 149, 139–153.
- Hamey, F.K.; Nestorowa, S.; Kinston, S.J.; Kent, D.G.; Wilson, N.K.; Gottgens, B. Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc. Natl. Acad. Sci. USA 2017, 114, 5822–5829.
- Schwab, J.D.; Ikonomi, N.; Werle, S.D.; Weidner, F.M.; Geiger, H.; Kestler, H.A. Reconstructing Boolean network ensembles from single-cell data for unraveling dynamics in the aging of human hematopoietic stem cells. Comput. Struct. Biotechnol. J. 2021, 19, 5321–5332.
- Herault, L.; Poplineau, M.; Duprez, E.; Remy, E. A novel Boolean network inference strategy to model early hematopoiesis aging. bioRxiv 2022.

