Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | yuanxi mo | -- | 1966 | 2022-10-17 17:55:07 | | | |
| 2 | Beatrix Zheng | + 7 word(s) | 1973 | 2022-10-18 08:04:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mo, Y.; Feng, Y.; Huang, W.; Tan, N.; Li, X.; Jie, M.; Feng, T.; Jiang, H.; Jiang, L. Role of Liquid–Liquid Phase Separation in Cardiovascular Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/29720 (accessed on 13 June 2026).
Mo Y, Feng Y, Huang W, Tan N, Li X, Jie M, et al. Role of Liquid–Liquid Phase Separation in Cardiovascular Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/29720. Accessed June 13, 2026.
Mo, Yuanxi, Yuliang Feng, Wei Huang, Ning Tan, Xinyi Li, Minwen Jie, Tong Feng, Hao Jiang, Lei Jiang. "Role of Liquid–Liquid Phase Separation in Cardiovascular Diseases" Encyclopedia, https://encyclopedia.pub/entry/29720 (accessed June 13, 2026).
Mo, Y., Feng, Y., Huang, W., Tan, N., Li, X., Jie, M., Feng, T., Jiang, H., & Jiang, L. (2022, October 17). Role of Liquid–Liquid Phase Separation in Cardiovascular Diseases. In Encyclopedia. https://encyclopedia.pub/entry/29720
Mo, Yuanxi, et al. "Role of Liquid–Liquid Phase Separation in Cardiovascular Diseases." Encyclopedia. Web. 17 October, 2022.
Copy Citation
Liquid–liquid phase separation (LLPS) is a biochemical process in cells that can drive proteins, RNA, and other molecules to concentrate into droplets. These droplets do not have a lipid membrane but rather exist as distinct organelles relative to the surrounding environment, and act as biochemical reaction chambers. Significant progress has been made in the study of LLPS, especially in the neurodegenerative disease, cancer, and virology fields, but little is known about LLPS in cardiovascular disease (CVD).
liquid–liquid phase separation
cardiovascular disease
membrane-free organelle
1. Cardiovascular Disease (CVD) and Intrinsically Disordered Proteins
Inherently disordered regions of proteins do not adopt a fixed tertiary structure, which contributes to the formation of multivalent interactions or misfolding in liquid–liquid phase separation (LLPS). Multivalent interactions are various non-covalent interactions formed when two molecules are combined. The IDRs of proteins are important factors driving phase separation. When proteins aggregate, an IDR undergoes a disorder-to-order transition [1] When all lysine residues in DDX3X-IDR1 were mutated to glutamine, a decrease in turbidity during LLPS in vitro was observed [2]. This showed that IDRs have an important role in phase separation.
Currently, there are more than 15,000 know proteins with an IDR. The IDRs can help proteins recognize and accelerate their interaction with other proteins and carry out alternative splicing, PTM, protein fusion, and insertion or deletion functions. Through database analysis of 487 proteins whose sequences were extracted from Swiss-Prot, 198 disordered regions among 101 proteins were predicted, and CVD-related proteins with disordered regions were very abundant [3]. Among these proteins, PDE4D may be involved in the occurrence of stroke through atherosclerosis. According to predictions, both the N-terminus and C-terminus of PDE4D contain disordered regions. The N-terminus participates in functions, such as phosphorylation and multimer formation, and the C-terminus is mainly related to dimer formation. Disordered regions can bind the SH3 domains of certain proteins, such as the Src family tyrosyl kinases lyn, fyn, and src. Furthermore, the SH3-binding domain of fyn kinase can bind proteins to induce LLPS [4]. Additionally, PDE4D may also interact with the SH3-binding domain of the kinase Fyn and induce phase separation.
2. LncRNAs-Associated Phase Separation in CVD
As non-coding RNAs, lncRNAs regulate epigenetic, transcriptional, and translation processes in organisms. Interestingly, lncRNAs can form the core of the separated droplets (Figure 1a). Furthermore, SGs often use untranslated mRNA as a scaffold and nucleosomes in the nucleus cluster with lncRNAs or pre-mRNA scaffolds [5]. Architectural RNAs (ArcRNAs) have a key role in particle assembly. The currently known arcRNAs are primarily mRNAs, and only five lncRNAs can act as arcRNAs, as follows: (1) the cores of nucleosome paraspeckles contain NEAT1, which mainly inhibits apoptosis and induces antiviral genes during viral infection and pregnancy establishment [6]. (2) Amyloid (amyloid bodies) uses IGS as its core and function to repair proteins in the body and regulate ribosome formation. (3) Satellite III in nuclear SGs is mainly involved in the separation of RBPs and transcription factors. (4) A heat shock RNA (hsr-omega) in omega speckles in Drosophila melanogaster is involved in normal development [7]. (5) Saccharomyces cerevisiae meiRNA in the Mei2 site is involved in the process of meiosis [8]. At the same time, lncRNA can also promote the phase separation of other proteins and recruit more proteins into the droplets, which also shows that lncRNA has an important role in phase separation [9].
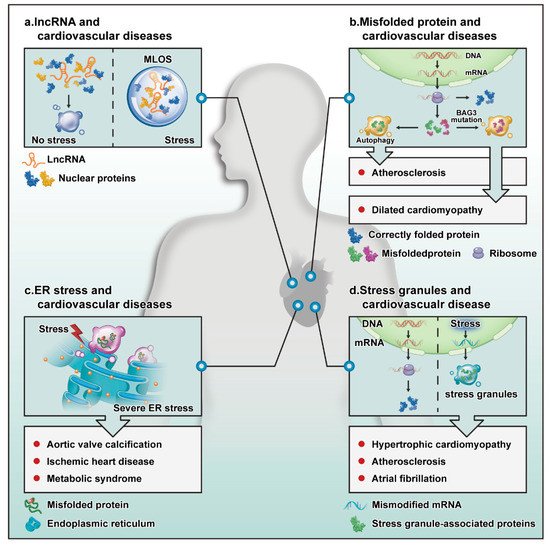
Figure 1. Potential role of LLPS in the cardiovascular system. (a) When stress occurs, lncRNAs become the core of droplets formed by liquid–liquid separation. (b) A correctly translated protein (blue) will perform its normal biological activities. Some misfolded proteins (green) are eliminated by autophagy to protect against atherosclerosis. Mutated or excessively produced proteins (red) accumulate, causing DCM. (c) The accumulation of a large number of misfolded proteins in the endoplasmic reticulum leads to endoplasmic reticulum stress, which can affect aortic valve calcification, ischemic heart disease, and metabolic syndrome. (d) Mistranscribed or modified mRNAs are eliminated by SG formation in the cytoplasm. This process can affect the progression of hypertrophic cardiomyopathy, atherosclerosis, and atrial fibrillation. Abbreviations are as follows: ER stress, endoplasmic reticulum stress; MLOs, membrane-free organelles.
Many lncRNAs have been reported to be closely related to CVD [10][11], and these relationships are usually related to the sponge function of microRNAs. So far, these lncRNAs have not been reported to undergo phase separation in cardiovascular disease. These lncRNAs may also help the formation of phase separation cores in cardiovascular diseases and may act as auxiliary tools to promote the formation of other phase separations. Whether these lncRNAs can also be used as arcRNAs to regulate downstream genes is worth investigating. Mannen et al. and Chujo et al. explored two methods to identify new nuclear bodies, namely RNase sensitivity screening and transcriptome screening of semi-extractable RNA, respectively [12][13]. These two methods can identify lncRNAs to assist in stent formation in CVDs.
3. Protein Misfolding in Cardiomyopathy
Protein folding is an important step by which proteins adopt a three-dimensional tertiary structure. To ensure normal operation in the body, proteins must be correctly folded to carry out their normal biological roles. Misfolded proteins are eliminated through the autophagy system. When regulation of protein expression or degradation pathways fails and degradation pathways are destroyed, proteins accumulate, and pathological changes occur. Interestingly, proteins that are not eliminated due to failure of the autophagy system may cause proteasome disease through phase separation (Figure 1b). In FTD and ALS, proteins accumulate due to targeted damage to degradation pathway proteins, such as FUS and TDP-43 [14][15]. Proteins may incorrectly accumulate for the following reasons: (1) autophagy and protein quality control (PQC) disorder can cause incorrect protein accumulation. When PQC proteins mutate, misfolded proteins cannot be cleared and accumulate, resulting in phase separation. For example, when BAG3 is deleted, or a residue in the BAG domain is mutated from glutamate to lysine (E455K), the interaction between BAG3 and HSP70 is reduced, thus, decreasing the stability of the PQC system [16]. (2) Mutated proteins cannot be eliminated by autophagy. The RNPs can cause protein aggregation through LLPS. Pathological aggregation is often caused by structural misfolding due to protein mutation [17]. (3) The mislocalization of proteins is caused by protein mutations, e.g., the FUS protein accumulates in ALS because most missense point mutations in FUS are concentrated on fragments that encode the nuclear localization sequence at the C-terminus. These mutations affect the nuclear localization of FUS and cause FUS to aggregate in the cytoplasm [15].
Recent studies have shown a close relationship between dilated cardiomyopathy (DCM) and protein mutation. For example, homozygous disruption of the Bag3 gene causes the fulminant form of DCM, and mutated CRYAB, which encodes α B-crystallin, was detected in patients with DCM [18][19]. A recent study showed a phase separation phenomenon caused by abnormal RNP particle accumulation in DCM caused by RBM20 mutation [20]. This also revealed a certain association between protein mutation and phase separation. It is known that BAG3, a multidomain chaperone protein, is a component of the HSP70-BAG3 complex. In addition, as an important signal transduction node, BAG3 is part of the Hippo-2 signal transduction complex in the Hsp70–Bag3–LATS1 pathway, which regulates protein aggregation [21]. Interestingly, the mutation of BAG3 can cause BAG3 to accumulate and HSP70 to aggregate [22]. This aggregation is induced because BAG3–HSP70–HSPB, a key complex, acts as a retrograde transport in autophagy in normal cells to promote the isolation and removal of irreversibly misfolded proteins in the PQC system. The mutation of BAG3 destroys the complex and prevents cells from carrying out normal PQC, resulting in misfolded protein accumulation and protein-related diseases.
4. The Cardiovascular System and Endoplasmic Reticulum Stress
Endoplasmic reticulum stress refers to the destruction of endoplasmic reticulum homeostasis and the accumulation of unfolded or misfolded proteins in the endoplasmic reticulum, leading to dysfunction of the endoplasmic reticulum and affecting cell functions, such as cell cycle regulation [23]. Atherosclerosis and ischemic heart disease are common CVDs and the main causes of myocardial ischemia. Endoplasmic reticulum stress may be an important mechanism for the further aggravation of myocardial ischemia. A recent study showed that the inhibition of endoplasmic reticulum stress could reduce the hypertrophy of cardiomyocytes and cardiomyocyte damage [24].
Wang et al. demonstrated that aortic valve calcification caused by hypercholesterolemia is related to endoplasmic reticulum stress in animal experiments [25]. It has also been reported that endoplasmic reticulum stress can downregulate HDAC6 and promote aortic valve calcification [26], which may be because the endoplasmic reticulum is an important intracellular calcium reservoir in cells. When endoplasmic reticulum stress occurs, endoplasmic reticulum dysfunction leads to disordered calcium regulation and aortic valve calcification (Figure 1c).
In addition, endoplasmic reticulum stress is related to metabolism. A high-fat diet may cause excessive production of oxidative free radicals through high levels of low-density lipoproteins (LDLs), destroying the homeostasis of the endoplasmic reticulum and causing endoplasmic reticulum stress [27]. A high-sugar diet can also damage endothelial cell function through endoplasmic reticulum stress. All these results prove the close relationship between endoplasmic reticulum stress and CVD.
5. Atherosclerosis and Autophagy
Phase separation plays an important role in promoting the formation of autophagosomes and the process of autophagy (Figure 1b) [28]. Atherosclerotic heart disease is a CVD with a high incidence, and the accumulation of abnormal protein promotes the progression of atherosclerosis and heart failure; therefore, the removal of these organelles and proteins by autophagy exerts protective effects against atherosclerosis. Autophagy has dual effects on the progression of atherosclerosis. Excessive autophagy or a delay in the macrophage autophagy response will aggravate the occurrence of atherosclerosis [29]. The ATG family genes are autophagy-related genes that play an important role in vascular regulation. When the ATG5 gene was knocked out, atherosclerosis in mice was aggravated [30]. Recent studies have also found that this gene and other ATG family genes can regulate protein autophagy through phase separation [31]. Therefore, the researchers speculate that autophagosomes are formed through phase separation, which recruits abnormal proteins to autophagosomes to achieve degradation, thereby affecting the process of atherosclerosis. In the future, exploring the role of phase separation in atherosclerosis will help delay the progression of the disease.
6. Atherosclerosis and SGs
Stress granules, particles that form in response to stress, are involved in regulating CVD (Figure 1d). For example, the accumulation of SGs can be observed in VSMCs and macrophages and affects the process of atherosclerosis [32]. Oxidized LDL (oxLDL) promotes the formation of such stress particles. As the first barrier in blood vessels, endothelial cells are most susceptible to stress caused by atherosclerotic stimulation, such as the modification of LDL levels and inducers of mitochondrial and oxidative stress, making it possible to explore the formation of pressure particles in endothelial cells.
Atrial fibrillation is the most common clinical arrhythmia and is also an important culprit of strokes caused by cardiovascular and cerebrovascular diseases. Enhanced cardiomyocyte activity is considered a strong stimulus for atrial fibrillation, and the heart may form SGs in response to such stimulation. The production of SGs was observed in primary cardiomyocytes and HL-1 cells after 1 h of pacing [33], which may be due to the excessive activity of cardiomyocytes that increases the oxidative stress of the myocardium and induces the production of SGs.
Hypertrophic cardiomyopathy is a genetic mutation-related disease. Some of these proteins, such as CALR3, NEXN, and TPM1, are also contained in SGs, which indicates that these proteins may regulate hypertrophic cardiomyopathy through phase separation. These proteins may have a role in the disease process through phase separation. For example, NEXN, a key heart-specific Z disc protein, can protect the Z disc from mechanical trauma [34]. Recent research shows that mutations in this protein could cause hypertrophic cardiomyopathy in zebrafish, and mutations in the corresponding gene can cause DCM in humans. The NEXN protein contains two actin-binding domains (ABDs) and a coiled-coil (CC) domain, and N-terminal ABD and CC domain mutants have been observed. These mutants exhibit local accumulation in the cytoplasm, while the ABD fragment from wild-type NEXN accumulates in the nucleus [35]. Thus, the mutation of NEXN causes an error in its localization, similar to the effect of FUS mutation.
References
- Wei, M.T.; Elbaum-Garfinkle, S.; Holehouse, A.S.; Chen, C.C.; Feric, M.; Arnold, C.B.; Priestley, R.D.; Pappu, R.V.; Brangwynne, C.P. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 2017, 9, 1118–1125.
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019, 15, 51–61.
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry 2006, 45, 10448–10460.
- Amaya, J.; Ryan, V.H.; Fawzi, N.L. The SH3 domain of Fyn kinase interacts with and induces liquid-liquid phase separation of the low-complexity domain of hnRNPA2. J. Biol. Chem. 2018, 293, 19522–19531.
- Yamazaki, T.; Nakagawa, S.; Hirose, T. Architectural RNAs for Membraneless Nuclear Body Formation. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 227–237.
- Yamazaki, T.; Souquere, S.; Chujo, T.; Kobelke, S.; Chong, Y.S.; Fox, A.H.; Bond, C.S.; Nakagawa, S.; Pierron, G.; Hirose, T. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 2018, 70, 1038–1053.e7.
- Singh, A.K.; Lakhotia, S.C. Dynamics of hnRNPs and omega speckles in normal and heat shocked live cell nuclei of Drosophila melanogaster. Chromosoma 2015, 124, 367–383.
- Mukherjee, K.; Futcher, B.; Leatherwood, J. mmi1 and rep2 mRNAs are novel RNA targets of the Mei2 RNA-binding protein during early meiosis in Schizosaccharomyces pombe. Open Biol. 2018, 8, 180110.
- Wu, M.; Xu, G.; Han, C.; Luan, P.F.; Xing, Y.H.; Nan, F.; Yang, L.Z.; Huang, Y.; Yang, Z.H.; Shan, L.; et al. lncRNA SLERT controls phase separation of FC/DFCs to facilitate Pol I transcription. Science 2021, 373, 547–555.
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583.
- Monteiro, J.P.; Bennett, M.; Rodor, J.; Caudrillier, A.; Ulitsky, I.; Baker, A.H. Endothelial function and dysfunction in the cardiovascular system: The long non-coding road. Cardiovasc. Res. 2019, 115, 1692–1704.
- Mannen, T.; Yamashita, S.; Tomita, K.; Goshima, N.; Hirose, T. The Sam68 nuclear body is composed of two RNase-sensitive substructures joined by the adaptor HNRNPL. J. Cell Biol. 2016, 214, 45–59.
- Chujo, T.; Yamazaki, T.; Kawaguchi, T.; Kurosaka, S.; Takumi, T.; Nakagawa, S.; Hirose, T. Unusual semi-extractability as a hallmark of nuclear body-associated architectural noncoding RNAs. EMBO J. 2017, 36, 1447–1462.
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363.
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507.
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200.
- Thelen, M.P.; Kye, M.J. The Role of RNA Binding Proteins for Local mRNA Translation: Implications in Neurological Disorders. Front. Mol. Biosci. 2019, 6, 161.
- Domínguez, F.; Cuenca, S.; Bilińska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M.; et al. Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481.
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386.
- Schneider, J.W.; Oommen, S.; Qureshi, M.Y.; Goetsch, S.C.; Pease, D.R.; Sundsbak, R.S.; Guo, W.; Sun, M.; Sun, H.; Kuroyanagi, H.; et al. Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat. Med. 2020, 26, 1788–1800.
- Klimek, C.; Jahnke, R.; Wördehoff, J.; Kathage, B.; Stadel, D.; Behrends, C.; Hergovich, A.; Höhfeld, J. The Hippo network kinase STK38 contributes to protein homeostasis by inhibiting BAG3-mediated autophagy. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1556–1566.
- Meister-Broekema, M.; Freilich, R.; Jagadeesan, C.; Rauch, J.N.; Bengoechea, R.; Motley, W.W.; Kuiper, E.; Minoia, M.; Furtado, G.V.; van Waarde, M.; et al. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat. Commun. 2018, 9, 5342.
- Lee, D.; Hokinson, D.; Park, S.; Elvira, R.; Kusuma, F.; Lee, J.M.; Yun, M.; Lee, S.G.; Han, J. ER Stress Induces Cell Cycle Arrest at the G2/M Phase Through eIF2α Phosphorylation and GADD45α. Int. J. Mol. Sci. 2019, 20, 6309.
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658.
- Cai, Z.; Li, F.; Gong, W.; Liu, W.; Duan, Q.; Chen, C.; Ni, L.; Xia, Y.; Cianflone, K.; Dong, N.; et al. Endoplasmic reticulum stress participates in aortic valve calcification in hypercholesterolemic animals. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2345–2354.
- Fu, Z.; Li, F.; Jia, L.; Su, S.; Wang, Y.; Cai, Z.; Xiang, M. Histone deacetylase 6 reduction promotes aortic valve calcification via an endoplasmic reticulum stress-mediated osteogenic pathway. J. Thorac. Cardiovasc. Surg. 2019, 158, 408–417.e2.
- Li, T.; Jiang, S.; Lu, C.; Hu, W.; Ji, T.; Han, M.; Yang, Y.; Jin, Z. Snapshots: Endoplasmic Reticulum Stress in Lipid Metabolism and Cardiovascular Disease. Curr. Issues Mol. Biol. 2018, 28, 14–28.
- Noda, N.N.; Wang, Z.; Zhang, H. Liquid-liquid phase separation in autophagy. J. Cell Biol. 2020, 219, e202004062.
- Zhang, J.; Ma, C.R.; Hua, Y.Q.; Li, L.; Ni, J.Y.; Huang, Y.T.; Duncan, S.E.; Li, S.; Gao, S.; Fan, G.W. Contradictory regulation of macrophages on atherosclerosis based on polarization, death and autophagy. Life Sci. 2021, 276, 118957.
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553.
- Fujioka, Y.; Alam, J.M.; Noshiro, D.; Mouri, K.; Ando, T.; Okada, Y.; May, A.I.; Knorr, R.L.; Suzuki, K.; Ohsumi, Y.; et al. Phase separation organizes the site of autophagosome formation. Nature 2020, 578, 301–305.
- Herman, A.B.; Silva Afonso, M.; Kelemen, S.E.; Ray, M.; Vrakas, C.N.; Burke, A.C.; Scalia, R.G.; Moore, K.; Autieri, M.V. Regulation of Stress Granule Formation by Inflammation, Vascular Injury, and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2014–2027.
- Dong, G.; Liang, F.; Sun, B.; Wang, C.; Liu, Y.; Guan, X.; Yang, B.; Xiu, C.; Yang, N.; Liu, F.; et al. Presence and function of stress granules in atrial fibrillation. PLoS ONE 2019, 14, e0213769.
- Hassel, D.; Dahme, T.; Erdmann, J.; Meder, B.; Huge, A.; Stoll, M.; Just, S.; Hess, A.; Ehlermann, P.; Weichenhan, D.; et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 2009, 15, 1281–1288.
- Wang, H.; Li, Z.; Wang, J.; Sun, K.; Cui, Q.; Song, L.; Zou, Y.; Wang, X.; Liu, X.; Hui, R.; et al. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2010, 87, 687–693.
More
Information
Subjects:
Anatomy & Morphology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
987
Revisions:
2 times
(View History)
Update Date:
18 Oct 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No