Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | FERNANDO CARRILLO HERMOSILLA | -- | 11751 | 2022-09-27 10:06:21 | | | |
| 2 | Camila Xu | -6334 word(s) | 5417 | 2022-10-18 03:34:23 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Carrillo-Hermosilla, F.; Fernández-Galán, R.; Ramos, A.; Elorriaga, D. Organometallic Chemistry of Guanidines. Encyclopedia. Available online: https://encyclopedia.pub/entry/29654 (accessed on 23 July 2026).
Carrillo-Hermosilla F, Fernández-Galán R, Ramos A, Elorriaga D. Organometallic Chemistry of Guanidines. Encyclopedia. Available at: https://encyclopedia.pub/entry/29654. Accessed July 23, 2026.
Carrillo-Hermosilla, Fernando, Rafael Fernández-Galán, Alberto Ramos, David Elorriaga. "Organometallic Chemistry of Guanidines" Encyclopedia, https://encyclopedia.pub/entry/29654 (accessed July 23, 2026).
Carrillo-Hermosilla, F., Fernández-Galán, R., Ramos, A., & Elorriaga, D. (2022, October 17). Organometallic Chemistry of Guanidines. In Encyclopedia. https://encyclopedia.pub/entry/29654
Carrillo-Hermosilla, Fernando, et al. "Organometallic Chemistry of Guanidines." Encyclopedia. Web. 17 October, 2022.
Copy Citation
Guanidines, nitrogen-rich compounds, appear as one such potential alternatives as ligands or proligands. In addition to occurring in a plethora of natural compounds, and thus in compounds of pharmacological use, guanidines allow a wide variety of coordination modes to different metal centers along the periodic table, with their monoanionic chelate derivatives being the most common.
guanidine
organometallics
catalysis
materials
1. Introduction
Guanidines, Y-shaped compounds of general formula R1N=C(NR2R3)(NR4R5) (R1–5=H, alkyl, aryl) (Figure 1), are very attractive organic molecules in different branches of Chemistry, exploiting their highly basic character [1][2] or the stability of their guanidinium cations, in which the positive charge could be delocalized over the three nitrogen atoms, and which are involved in many biological processes. For example, several enzymes containing the amino acid arginine take advantage of the presence of the side chain functional group guanidinium for the fixation of diverse substrates [3][4][5][6][7][8][9]. Similarly, the facility to form very stable hydrogen bonds allows the use of guanidines as organocatalysts in reactions, such as Diels–Alder reactions, addition reactions, CO2 capture and transformation to valuable products or the ring-opening polymerizations of lactones and lactides, even with the adequate control of the enantioselectivity [10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32]. It should be noted that the expected donor character of these derivatives and their anionic forms, the guanidinates, has led to a new and growing class of metal complexes. Although there are a significant number of examples where guanidines act as neutral ligands, the monoanionic forms exceed neutral or dianionic forms in examples, the N,N’-chelating being by far the most widely encountered coordination mode.
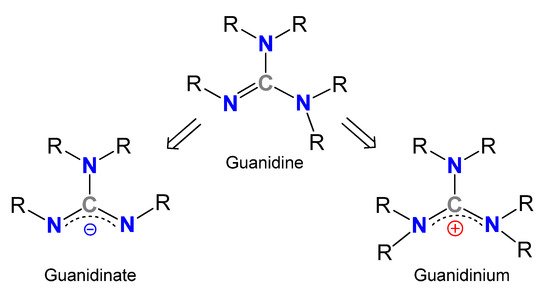
Figure 1. General structures of guanidines, guanidinates and guanidinium cations.
The main feature of these chelating monoanionic guanidinato ligands is the electronic flexibility in coordination with metal centers due to the large electronic delocalization through its structure (Figure 2). This in turn provides stabilization to a wide variety of metal centers throughout the periodic table, in different oxidation states.
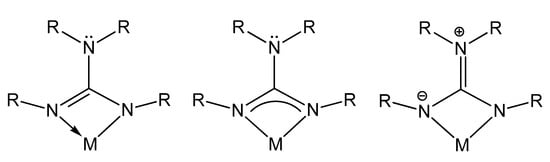
Figure 2. Electronic delocalization in guanidinato ligands.
It is noteworthy that the great interest in the study of these compounds provides a broad palette of synthetic methods, involving both stoichiometric and catalytic processes [33][34][35][36][37].
Since metallic compounds based on these ligands were first reported in 1970 [38], they have shown great promise for applications in catalysis and materials science. In particular, guanidinato ligands have been investigated as an interesting alternative to classical ligands, such as cyclopentadienyl. An appropriate modification of the steric properties of these ligands can also play an important role in the commented applications of these complexes.
2. Organometallic Chemistry of Guanidines
2.1. Main Group Complexes
Although, as mentioned above, the most common coordination mode of guanidinato ligands is the chelating k2-N,N’ form, in the case of bulky lithium guanidinates, additional metal–carbon bond interactions can occur due to the presence of aromatic groups. These M-arene interactions have also been found for other group 1 elements, which are used to obtain guanidinates of other metals, with the purpose of generating steric protection for these metal centers.
The deprotonation of the bulky guanidine ArNC(NCy2)N(H)Ar (Ar = 2,6-iPr2C6H3) with BunLi in THF led to the monomeric complex 1. Similarly, the deprotonation of ArNC(NiPr2)N(H)Ar with K[N(SiMe3)2] gave an unsolvated polymer, 2. whereas the arene-K interactions were close to η6- and the lithium compound showed an organometallic bonding of the type η3- (Figure 3) [39][40].
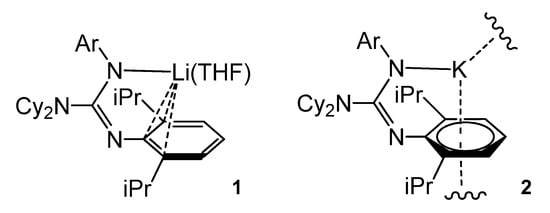
Figure 3. Lithium and potassium compounds with η6-arene coordination.
The polymeric structure is repeated in derivative 3 with methyl groups on the aromatic rings. The solid-state structures reveal that the guanidinato ligand in these compounds retains the typical Z-anti configuration occurring in this class of bulky guanidines (Figure 4).
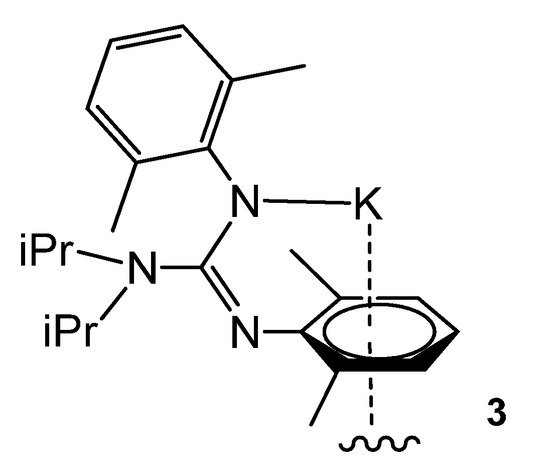
Figure 4. Polymeric potassium compound through η6-arene interactions.
The reaction of bulky carbodiimide (ArN)2C (Ar = 2,6-(diphenylmethyl)-4-tert-butylphenyl) with LiNCtBu2 in tetrahydrofuran gives the monomeric lithium guanidinato compound 4. Protonation and subsequent metalation allow the analogous compounds with K 5 and Cs 6 to be obtained (Figure 5). The solid-state structures of these complexes show that the alkali cations sit within a cavity formed from the peripheral phenyl rings of the diphenylmethyl substituents, which also provide additional support to the cations via intramolecular metal–arene interactions [41].
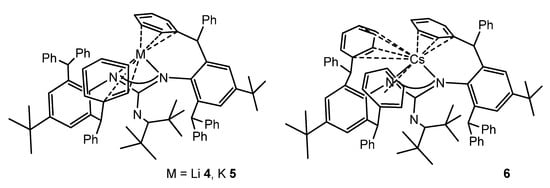
Figure 5. Alkali metal complexes exhibiting bond interactions with the aromatic substituents of the guanidinato ligands.
Other bulky guanidines, bearing 2,6-diisopropylphenyl (Dipp) group as substituent show, again, a coordination of the aromatic group to the metal center. This is the case in compounds 7–10 (Figure 6) [42].
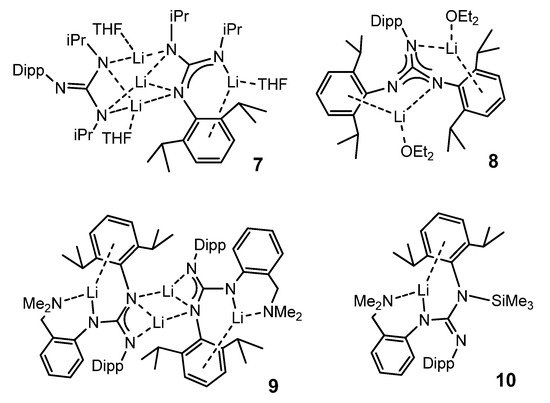
Figure 6. Alkali metal complexes exhibiting bond interactions with the aromatic substituents of the guanidinato ligands.
In the search for bimetallic systems with multiple metal-to-metal bonds, the reduction of a chromium guanidinate with KC8 led to a monomeric chromium (0) complex 11, in which this metal atom is sandwiched between two arene units of two guanidinato ligands. The coordination of the arene units is not limited to the chromium atom, but the same aromatic substituents also coordinate one potassium cation in a similar fashion. Furthermore, the guanidinato ligands in 11 act as amides coordinating the K atom (Figure 7) [43].
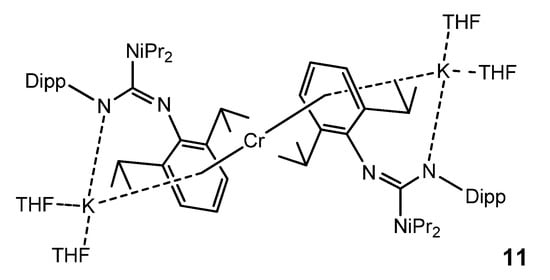
Figure 7. Monomeric chromium (0) bimetallic complex.
The organometallic derivatives of most of the group 2 elements have been described. Very bulky guanidinato ligands enable the stabilization of abnormally low oxidation states for some metals. A dimeric magnesium(I) guanidinato complex acts as a facile and selective two-center/two-electron reductant towards a series of unsaturated substrates. The reaction with CyNCNCy gives the doubly reduced product 12 (Figure 8) [44].
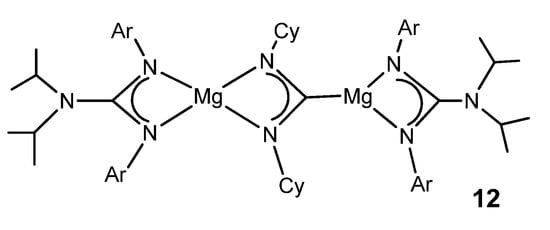
Figure 8. Magnesium magnesioamidinato complex.
The Schlenk equilibrium between the bis-guanidinato complex of calcium [Ca{(Cy)N–C[N(SiMe3)2]–N(Cy)}2·(Et2O)] and the benzyl derivative [Ca{α-(Me3Si)-o-(Me2N)benzyl}2·(THF)2] yields mainly the heteroleptic benzylcalcium complex 13 (Figure 9) [45]. This complex, like many others that will appear throughout the text, is an example of one of the main applications of the compounds described in this work: to act as catalysts, in this case, in the catalytic polymerization of olefins in particular.
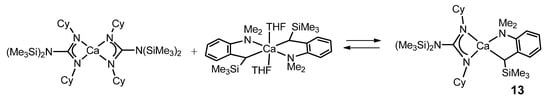
Figure 9. Heteroleptic benzylcalcium complex as the product of Schlenk equilibrium.
In the case of the heavier group 2 elements, finding organometallic derivatives with potential uses is often hampered by their low stability and tendency to form poorly soluble oligomeric species. This is a consequence of the large ionic radius of the strontium and barium metal centers. These problems can be addressed by employing suitable bulky ligands that limit this tendency to aggregate. For example, a combination of guanidinato ligands and Cp* (Cp*=C5Me5) allows dinuclear strontium and barium species to be obtained, where the guanidinato chelate ligands also act as bridges between the metal atoms (Figure 10) [46].
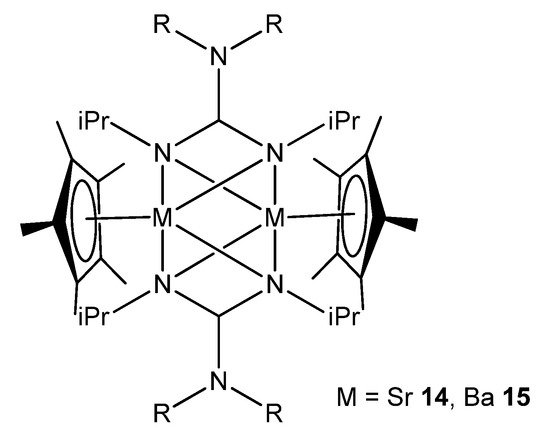
Figure 10. Dinuclear strontium and barium compounds with guanidinato chelate and bridging ligands.
As with some group 1 elements, in the case of the barium complex 16, electronic stabilization occurs, in addition to the steric protection of the guanidinato ligands, via interactions between the metal cation and the aromatic substituents of the ligands (Figure 11) [47].
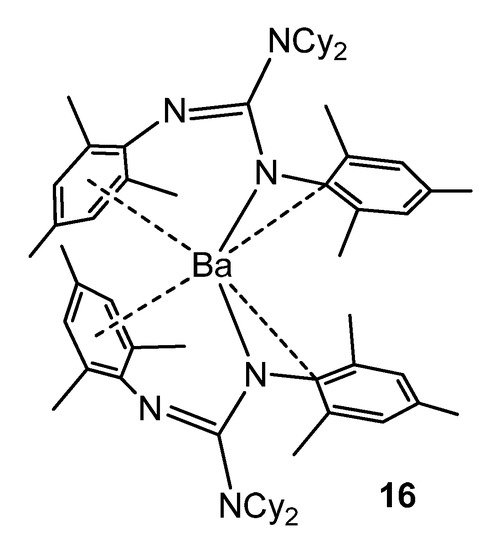
Figure 11. Electronic and steric stabilization of barium by aryl substituents in guanidinato ligands.
Although zinc is a d-block element, it is not considered a transition metal and is more appropriately considered as part of the main group. Interestingly, zinc organometallic guanidinates are scarce. The selectivity of the reaction between zinc dialkyls and guanidines is mediated by the identity of the guanidine substrate. The use of bis-guanidines with bulky aromatic substituents leads to the formation of dimer species, which have electron-deficient ethyl bridges common in the organometallic chemistry of the main group. These compounds are suitable precursors for obtaining stable zinc hydride derivatives, as they are active in the hydroboration and hydrosilylation of ketones (Figure 12) [48].
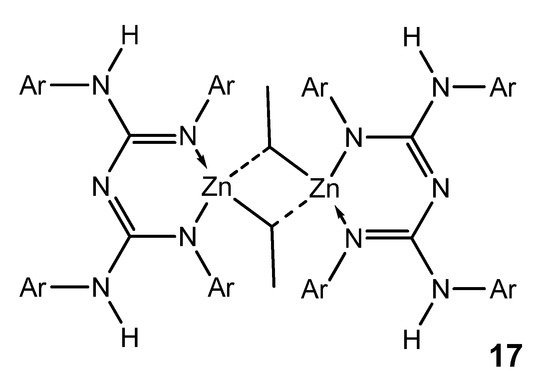
Figure 12. Bis-guanidianato alkyl zinc dimeric complex.
In the case of bicyclic guanidines, polynuclear systems can be achieved, where the more open projection of the orbitals of the nitrogen atoms of the amidine component of the ligand allows the formation of guanidinato bridges, as opposed to the formation of chelate systems (Figure 13) [49][50][51].
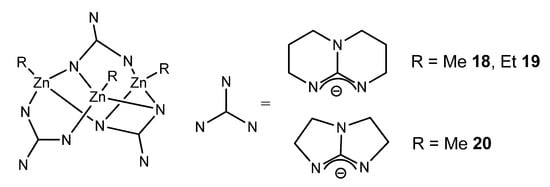
Figure 13. Bicyclic guanidinato complex acting as bridges in alkyl zinc complexes.
With less sterically demanding guanidines, it is also possible to obtain dimer species in which the guanidinato ligands act both as chelate ligands and as a bridge between two zinc nuclei (Figure 14) [52][53]. The resistance to the protonation of the remaining alkyl group is manifested in the fact that protonation with a phenol involves the preferential cleavage of a Zn–N bond, generating a zinc complex with a neutral guanidine ligand coordinated by the imine nitrogen.
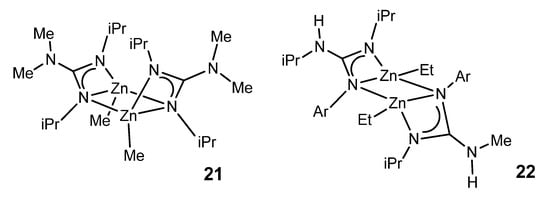
Figure 14. Guanidinato ligands acting both as chelate ligands and as a bridge between two zinc nuclei with boat and chair conformations.
The ethylzinc complexes of the type of 22, with asymmetrically coordinated guanidinato ligands, have been shown to be intermediates for the catalytic production of new trisubstituted guanidines by the direct addition of anilines to carbodiimides (Figure 15) [53].
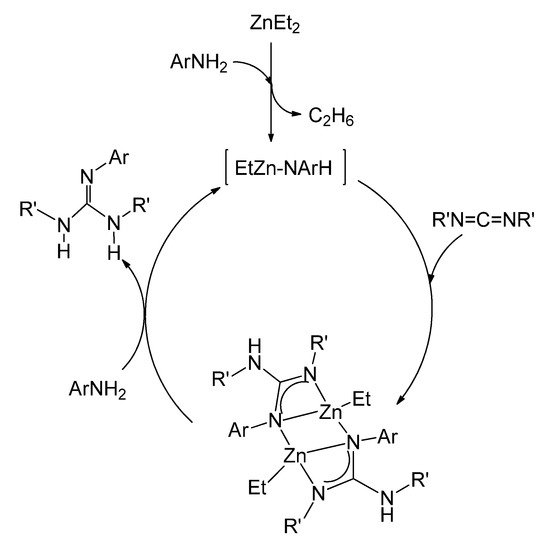
Figure 15. Catalytic guanylation of amines using ZnEt2 as a precatalyst.
There is the widespread organometallic chemistry of aluminum compounds with guanidinate ligands. This interest can be seen in the trend of the last decades in the search for catalysts based on Main Group metals, looking for catalytic systems less expensive and with less impact on the environment than traditional catalysts based on transition metals [54].
Thus, coordination and organometallic aluminum complexes with a well-defined structure are among the most interesting due to their Lewis acidic characteristics and their availability.
These aluminum guanidinate derivatives can be obtained both by metathesis of the ligand salt and an aluminum halide or by the protonolysis of alkylaluminums (Figure 16) [43][55][56][57][58][59].
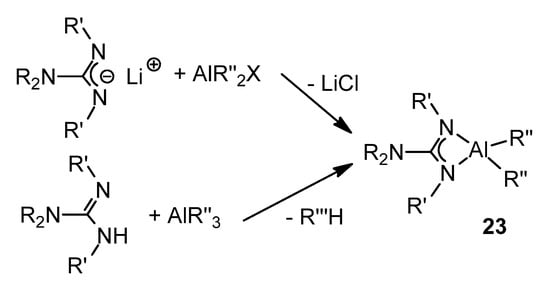
Figure 16. Synthesis of organoaluminum guanidinato complexes.
Depending on the type of guanidine, systems with chelating or bridging ligands can be obtained. As previously described for zinc [49][50][51], the arrangement of the electron pairs of the donor atoms in the bicyclic ligand makes a chelate-type coordination difficult (Figure 17).
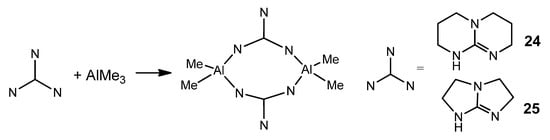
Figure 17. Bicyclic guanidinates acting as bridging ligands.
An alternative is the migratory insertion of carbodiimides into AlR2(NR’2)-type compounds. In fact, it has been shown that the amido groups are preferred to the alkyl or halide groups for migration [60].
These amido derivatives can be generated in situ and lead to the insertion of the carbodiimide to form the guanidinato species (Figure 18). It should be noted that this can occur during the catalytic process of addition of amines to carbodiimides to form new guanidines. The acid–base reaction between an aluminum alkyl and an N–H bond of the amine results in the formation of the amido species which, via nucleophilic addition to the carbodiimide, followed by protonolysis by another amine molecule of the guanidinato intermediate, gives rise to the corresponding guanidine [56][61][62].
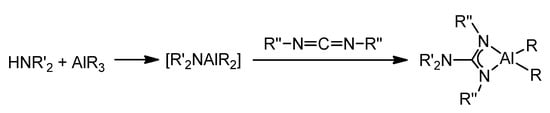
Figure 18. Insertion of carbodiimides into Al–N bonds of amido compounds.
The high kinetic inertness and thermodynamic stability provided by the guanidinato ligands to their complexes makes them very interesting precursors for the formation of transition metal–aluminum bonds by suitable reactions with organometallic anions (Figure 19) [63].
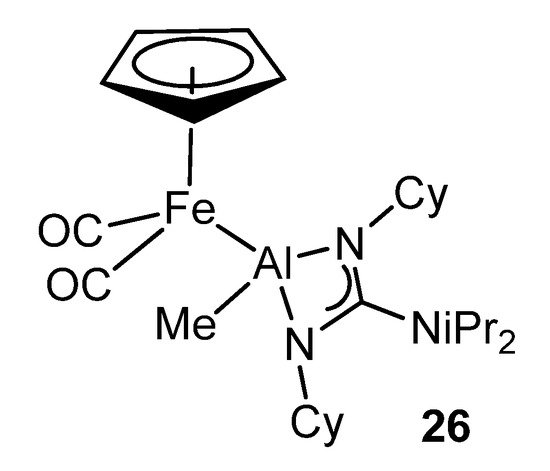
Figure 19. Bimetallic iron–aluminum compound.
An interesting example of how a guanidinato ligand is obtained occurs when alkylaluminums and a biscarbodiimide, or two carbodiimide molecules, react at high temperatures. Instead of the desired amidinate, after the insertion process of one of the carbodiimide groups, the reaction proceeds with the attack of the nitrogen of the formed amidinate to the electrophilic carbon atom of the second carbodiimide (Figure 20) [64][65].
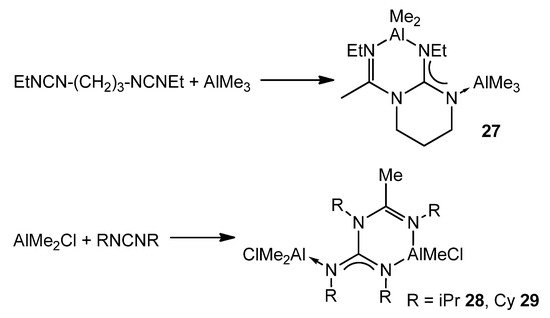
Figure 20. Double insertion of carbodiimides.
This reaction has also been observed for aluminum guanidinato compounds but depends on the nature of the substituents on the carbodiimide. Thus, [Al{k2-(NCy)(NPh)C(Net2)}Me2] reacts with CyN=C=NPh, but not with CyN=C=Ncy, to give rise to a complex similar to those of Figure 20 [66].
The potential of some of these organometallic complexes to act as catalysts is manifested in their ability to selectively reduce the carbonyl group of unsaturated aldehydes and ketones by the Meerwein–Ponndorf–Verley reaction [59][67].
In relation to ketone and aldehyde reduction, the feasibility of transforming some of these organometallic complexes into hydride derivatives should be highlighted. From bis-guanidines, mononuclear and dinuclear hydroboration or hydrosilylation catalysts can be obtained (Figure 21) [68][69].
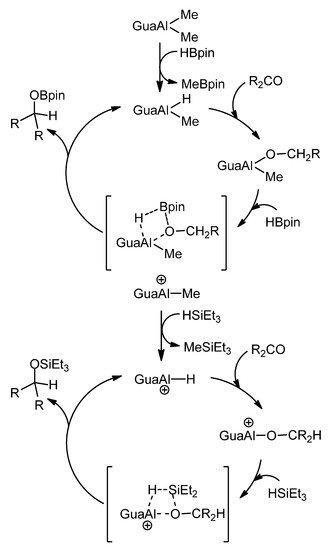
Figure 21. Hydroboration and hydrosilylation of ketones by aluminum hydride compounds.
Likewise, the dinuclear complexes of the type 30 (Figure 22) are efficient catalysts for the ring-opening polymerization of L-lactide and ε-caprolactone [70].
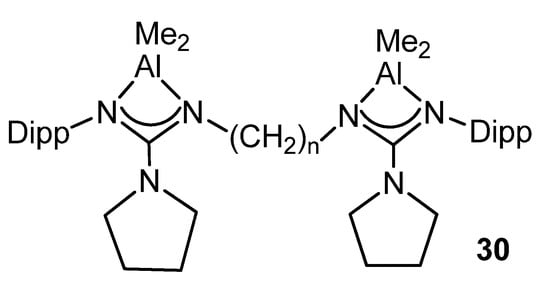
Figure 22. Dinuclear aluminum compounds, catalyst for ring-opening polymerization (n = 2, 3, 4).
The protonolysis of AlMe3 with trisubstituted arylguanidines allows to prepare mono- and dinuclear complexes, with different coordination modes. Through an appropriate choice of molar ratio and ligand structure, it is possible to achieve mononuclear complexes with the conventional chelate tetrahedral coordination, complexes with two AlMe2 moieties, or mononuclear with a rare pentacoordinated environment around the aluminum metal center (Figure 23). Some of these complexes are very active catalysts for the transformation of styrene oxide and CO2 into the corresponding cyclic carbonate [71].
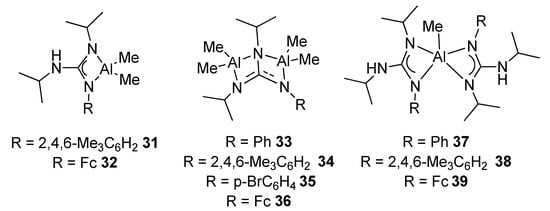
Figure 23. Mononuclear and dinuclear aluminum compounds with different coordination numbers.
The modification of guanidines with phosphane groups generates N-phosphanoguanidines, of general formula (HNR)(Ph2PNR)C(NAr). In the reaction with trimethylaluminum, phosphanoguanidinato derivatives were detected or isolated as the kinetic products of the reaction. Surprisingly, when the solutions of these organometallic complexes are heated, it results, selectively and quantitatively, in phosphanimine–amidinato compounds as the thermodynamic products of the process (Figure 24). A similar process was observed for other metals, such as Zn, Mg and Li [72][73].
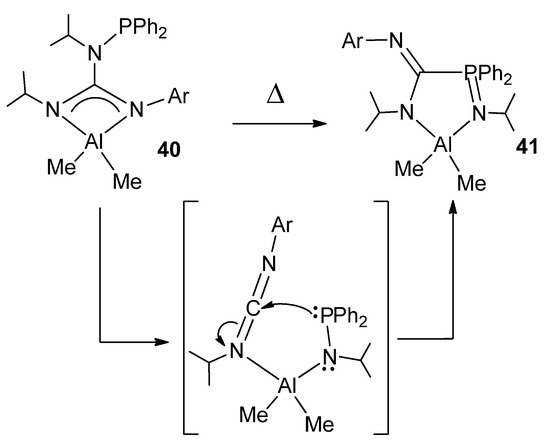
Figure 24. Rearrangement in phosphanoguanidinato derivatives.
Finally, as will be described later for other elements, because of its stability and volatility, organometallic aluminum guanidinates are potential precursors for the deposition of high-quality aluminum oxide by atomic layer deposition (ALD) [74].
Much less common are references to organometallic complexes of the other group 13 elements and other main group metals. These complexes of gallium, indium, thallium, tin and lead often display guanidinato ligands with substituents that complete the coordination sphere of the metal, such as aryl, amido, alkoxy or iminoacyl groups [75][76][77][78][79][80][81][82][83][84][85][86][87].
2.2. Transition Metal Complexes
Some of the alkali metal guanidinates described above were synthesized or prepared in situ as starting materials for reactions with transition metal halides and related precursors. Although the guanidinato derivatives of transition metals had already been described [38], Richeson’s group opened up the organometallic chemistry of metals of the early groups with this type of ligands. The alkylation of a symmetrically substituted guanidinato derivative of zirconium by metathesis reaction with PhCH2MgCl generates complex 42 (Figure 25). As with analogous classic complexes with cyclopentadienyl ligands, this compound exhibits a η2-type bond interaction of a benzyl ligand [88].
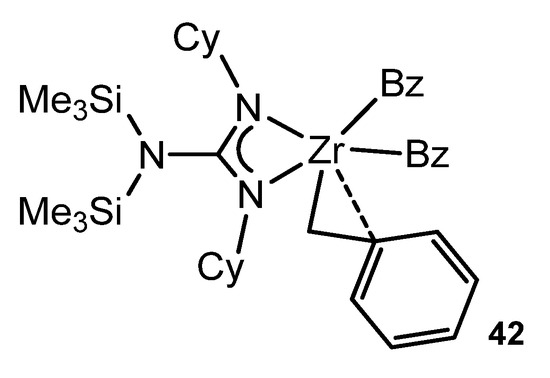
Figure 25. Zirconium complex with a η2-type bond interaction of a benzyl ligand.
Alternatively, the same complex could be accessed via a toluene elimination reaction between neutral guanidine and tetrabenzylzirconium. This reaction can also be extended to other alkyltitanium derivatives [89][90][91].
There are far fewer examples of complexes containing a guanidinato (2−) ligand than the monoanionic ligands mentioned above [92][93]. For example, the reaction of [Cp*ZrCl3] with the dianion [{(PhN)3C}Li2] gave the zwitterionic species [Cp*Zr{C(NPh)3}Cl2Li(Et2O)(THF)], a complex active in ethylene polymerization [92]. As for their monoanionic analogues, these doubly negatively charged ligands also exhibit chelate-type coordination. Fluxional behavior was found in solution, and this involves a syn/anti isomerization process of the guanidinato (2−) ligand [93].
The formation of mixed cyclopentadienyl zirconium guanidinato derivatives can also be achieved by a formal [2 + 2] cycloaddition reaction of carbodiimides to imides. These azaallyl-like ligands still exhibit the κ2-chelate coordination described above and not the possible κ3-coordination similar to that of the allyl ligands, due to the orbitals involved in both cases (Figure 26). These complexes enable exchange reactions with other carbodiimides under mild conditions, which extends the family of this type of derivatives [94][95].
Figure 26. Dianionic zirconium guanidinato complex.
This type of complex still presents an uncoordinated imino group with a lone pair that could participate in the coordination to Lewis acid centers. However, the more than likely delocalization of these electrons along the ligand prevents such reactivity [96].
Whereas the alkylation with MeLi of the derivative Cp(C(C(NiPr)2(NMe2))ZrCl2 leads to a metathesis process to form the metallocene derivative and the bis-guanidinate, the use of the Grignard reagent MeMgCl or, more conveniently, the protonation reaction of the derivative (C5H3(SiMe3)2)ZrMe3 with triisopropylguanidine allows the obtention of dialkyl derivatives with guanidinato ligands [94][95][97].
Following the similarity with the chemistry of metallocene derivatives, these neutral alkyl zirconium complexes with guanidinato ligands can be transformed into cationic derivatives by reaction with reagents, such as tris(pentafluorophenyl)borane, [Ph3C][B(C6F5]3]4 or Me3SiOTf (Figure 27).
Figure 27. Cationic derivatives of a bis-guanidinato zirconium compound.
The great influence of the geometry and the donor capacity of the guanidinato ligands on the reactivity of the complexes they form can be exemplified by studying the reactions of dialkyl derivatives with aryl isonitriles. The titanium or zirconium complexes of this class form intermediates, sometimes isolated, with η2-iminoacyl ligands for which subsequent transformations lead to vinylamido-type species, in the case of linked guanidinato ligands [98], while their unlinked analogues evolve towards imido or enediamido species (Figure 28) [99][100][101].
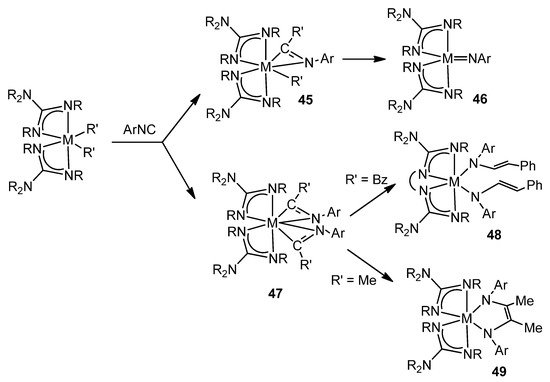
Figure 28. Aryl isonitriles insertion and the consecutive evolution of the iminoacyl groups.
Occasionally, the lithiated guanidine derivative does not generate the expected chelate complex observed in most of the compounds described. Instead, in complexes such as Cp2TiCl(k1-N=C(NMe2)2) 50, the guanidinate assumes an unusual binding mode, in the form of a monodentate ligand [102][103].
The metallacarborane complexes of zirconium and titanium can be obtained either by protonolysis with neutral guanidine or carbodiimide insertion in amido precursors. Titanium complex 51 is an effective catalyst for the addition of primary and secondary aliphatic and aromatic amines to carbodiimides with good functional group tolerance (Figure 29) [104][105].
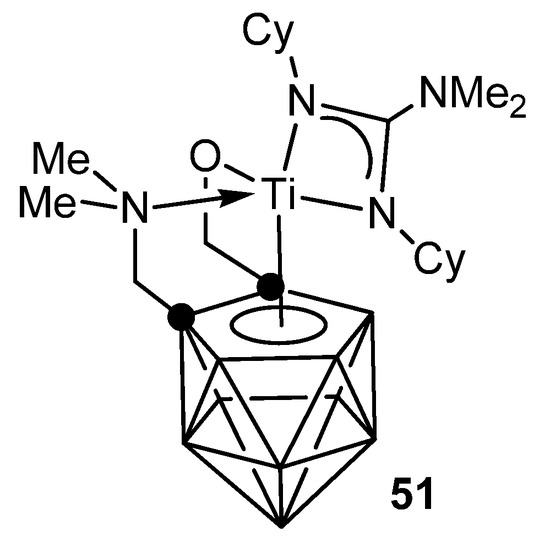
Figure 29. Metallocarborane complexes of titanium.
Using chiral modified 2-aminooxazolines gave rise to chiral half-sandwich complexes where the amino-oxazolinato group had undergone a ring opening and a migratory insertion of a dimethylamido ligand, providing a configurationally stable chiral-at-metal system (Figure 30) [106][107].
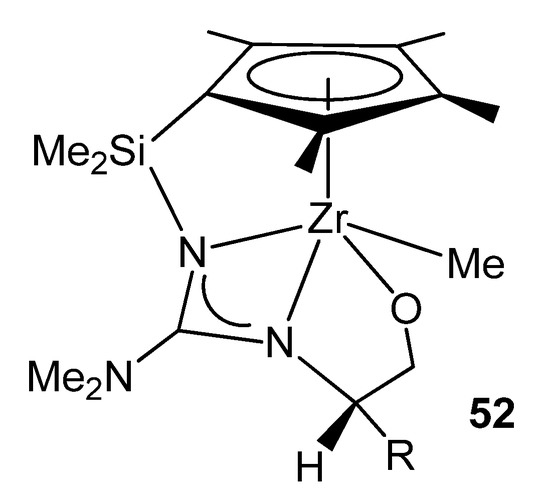
Figure 30. Chiral half-sandwich complexes with an oxazolinato group.
Although, as with metallocene derivatives, their application in catalytic processes makes the guanidinato complexes of group 4 metals very attractive compounds, these complexes have received interest as precursors to metal nitride or oxide thin films due to their nitrogen or carbon content, their potential to increase the volatility of the compound and their ability to stabilize the metal center due to their electronic flexibility. For example, thin films of carbonitrides or oxides of titanium, zirconium or hafnium can be obtained from guanidinato or mixed cyclopentadienyl guanidinato precursors. The above properties of these molecules satisfy the important requirements for their application in chemical deposition techniques, such as ALD or CVD [108][109][110][111].
As mentioned above, guanidinato ligands allow the stabilization of less common oxidation states for transition metals. Some mixed halide complexes of zirconium and hafnium can be reduced in the presence of N2 to form bimetallic complexes with a side-on-bridged dinitrogen molecule and proved to be very active toward both hydrosilylation and hydrogenation (Figure 31) [112].
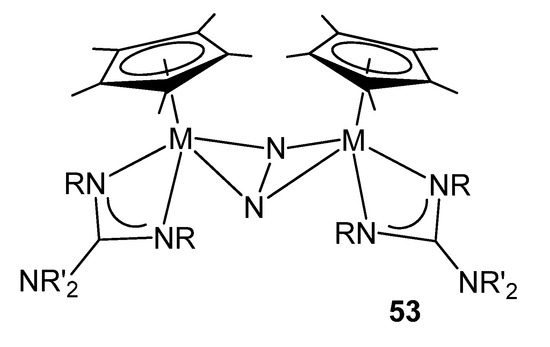
Figure 31. Coordination and activation of dinitrogen with group 4 guanidinato complexes.
The titanium (II) complex 54 (Figure 32), obtained by a two-electron reduction from a dichloride precursor, exhibits an arene–M bond interaction similar to that previously described for group 1 elements, such as lithium or potassium. This type of complexes allows catalytic hydrogenation by hydrogen transfer to monocyclic and polycyclic arenes, mimicking the reactivity of late transition metals, as well as demonstrating a great ability to reductively activate a wide range of substrates, such as ketones, azides, N2O or C–F bonds [113][114][115].
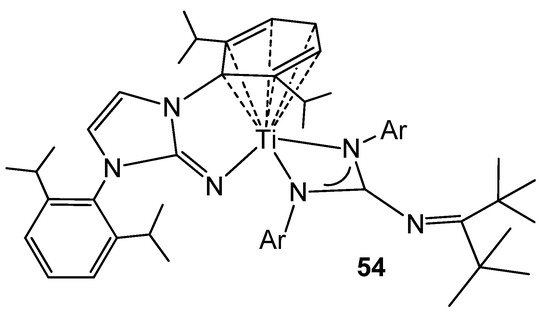
Figure 32. Arene–metal bond interaction in a titanium complex.
There is some parallelism between the organometallic chemistry of guanidinato complexes of groups 4 and 5. For example, the alkyl derivatives of niobium of the type 55 (Figure 33) show a η2-benzyl group in the solid state, and this is in rapid exchange with the other η1-benzyl ligand in solution. While the migratory insertion of tBuNC into the niobium–alkyl bonds gives rise to the corresponding bis(iminoacyl) derivatives, the insertion of an aryl isonitrile, XyNC (Xy = 2,6-Me2C6H3), into a niobium–benzyl bond results, as with some zirconium complexes [99], in the isomerization of an η2-iminoacyl group to a vinylamido-type one via a 1,2-hydrogen shift. The influence of the guanidinato ligand on this process is clear, since the same reaction does not take place starting from the precursor compound of the guanidinato complexes, the trialkylimido [NbBz3(NtBu)] (Bz = benzyl) [116][117].
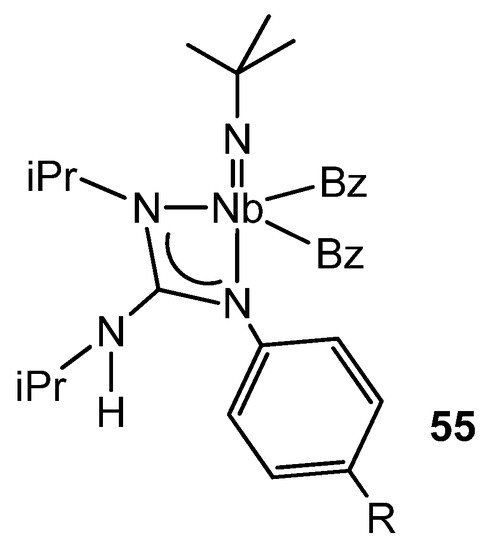
Figure 33. Asymmetric coordination of a guanidinato ligand in niobium complexes.
These dibenzyl complexes with guanidinato ligands derived from diisopropylcarbodiimide and an aromatic amine show an asymmetric coordination of the ligand via an alkylamino nitrogen atom and the arylimino nitrogen atom. Computational studies confirm this preference, and the results suggest that electronic factors prevail over steric factors. In fact, when a guanidine with three alkyl substituents, one of them different, was used for the synthesis, a mixture of isomeric complexes was formed with the coordinated ligands without any selectivity for any of the nitrogen atoms. In addition, these complexes were proposed as intermediates in the mechanism of the catalytic guanylation of anilines, using the complex [NbBz3(NtBu)] as a precatalyst [118].
Another example of the remarkable influence of the coordination of a guanidinato ligand on the reactivity on the coordination sphere of a metal is the process, in which an alkyl guanidine reacts with an iminocarbamoyl complex, which undergoes an easy cleavage of a C–N bond at room temperature (Figure 34) [119].
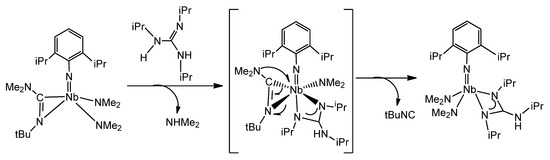
Figure 34. C–N activation mediated by a guanidine in an iminocarbamoyl compound.
This process contrasts with that observed when migratory insertion of aryl isonitriles into the Ta-NMe2 bonds of a previously formed guanidinato complex was undertaken [120]. In this case, the iminocarbamoyl species [Ta(NMe2)[C(NiPr)3][η2-(Me2N)C=N(2,6-Me2C6H3)]2] 56, which is stable, was formed.
As was the case for group 4 metals [112], guanidinato ligands stabilize organometallic tantalum species in a medium or low oxidation state [121][122][123]. For example, mixed cyclopentadienyl-guanidinato species of Ta(IV) give rise to N≡N bond cleavage to provide the bimetallic bis(μ-nitrido) complex, {Cp*Ta[N(iPr)C(NMe2)N(iPr)](μ-N)}2 57. The presence of the uncoordinated NMe2 group in the guanidinato ligand can serve to modulate the magnitude of the free energy barrier for N≡N bond cleavage.
Very recently, the participation of tantalum alkyl guanidinato derivatives of the type [Ta(hpp)2(CH2SiMe3)3] (hpp = 1,5,7-triazabicyclo [4.4.0]dec-5-ene) 58 has been studied as probable intermediates in the process of hydroaminoalkylation of terminal alkenes with a variety of secondary amine substrates to give substituted secondary amine products [124].
While the organometallic chemistry of group 6 derivatives is limited to a molybdenum complex, [(ArN)2MoMe{N(Cy)C[N(iPr)2]N(Cy)}] (Ar = 2,6-iPr2C6H3) 59 [125], and that of group 7 is non-existent, group 8 organometallic complexes with guanidine-derived ligands offer interesting structural and reactivity aspects.
Iron guanidinato (2−) complexes 60 and 61 (Figure 35) were obtained via a formal [2 + 2] cycloaddition of carbodiimide to imido precursors in high or low oxidation states [126][127].
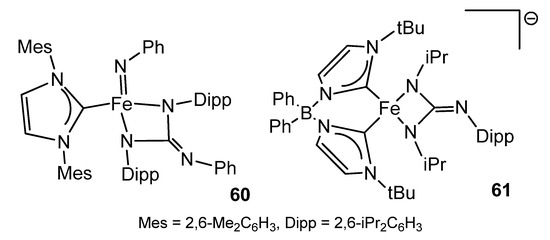
Figure 35. Iron dianionic guanidinato compounds.
Ruthenium witnesses the first reference of a complex in which the guanidinato ligand acts as a chelate. Indeed, complex 62 contains an η6-bonded aromatic ligand, a terminal chloride and the chelating triphenylguanidine anion. The structure of this compound shows, for the first time, a feature that is repeated in many guanidinato complexes: the angles around the central carbon of the guanidine ligand total 360° indicating the strict planarity of the CN3 core (Figure 36) [128].
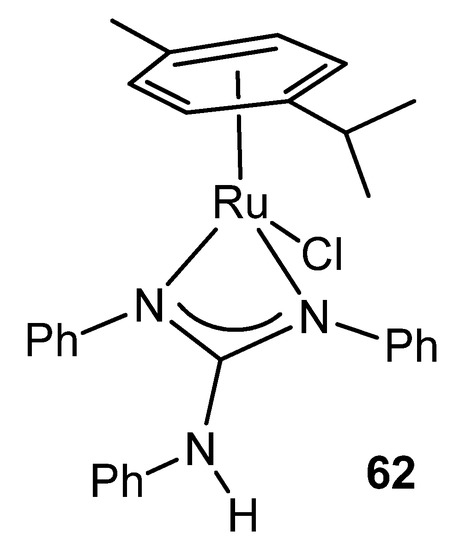
Figure 36. First example of a chelate guanidinato compound.
This coordination mode is present in other similar ruthenium complexes with both monoanionic and dianionic ligands [129][130][131][132][133].
It is also worth noting that, despite the prominent role of ruthenium complexes in organic synthesis, there are only a few examples of guanidinato complexes reported to date that have been explored as catalysts. Complexes similar to those described are excellent catalysts for the redox isomerization of allylic alcohols in the absence of base (Figure 37) [134].
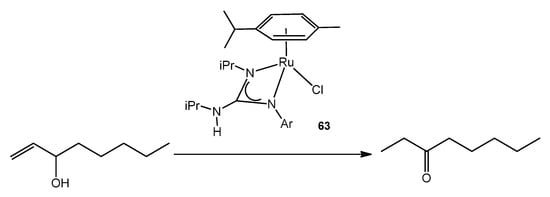
Figure 37. Ruthenium guanidinato complex acting as catalyst for allylic alcohol isomerization.
This catalytic process also takes place using ruthenium (IV) guanidinato complexes, namely [RuCl{κ2-C(NR)(NiPr)NHiPr}(η3:η3-C10H16)] 64 (Figure 38). This type of compounds are excellent precursors for the preparation of octahedral ruthenium (II) guanidinato complexes, mer-[RuCl{κ2-C(NR)(NiPr)-NHiPr}(CN-2,6-C6H3Me2)3] 65, through the reductive elimination of the ligand 2,7-dimethylocta-2,6-diene-1,8-diyl with high stereoselectivity [135].
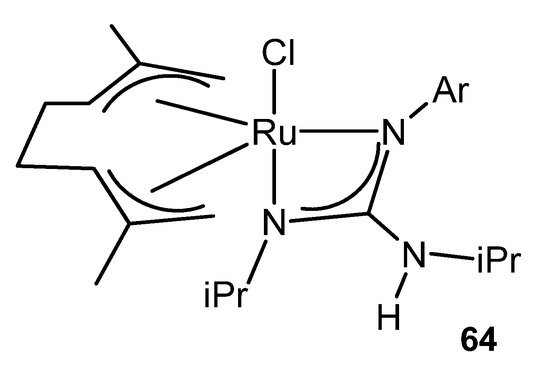
Figure 38. Ruthenium (IV) guanidinato complex [RuCl{κ2-C(NR)(NiPr)NHiPr}(η3:η3-C10H16)].
In contrast, less attention has been paid to osmium guanidinato complexes [129][136], although some examples were able to transform a wide variety of aldoximes selectively and catalytically into the corresponding nitriles (Figure 39).
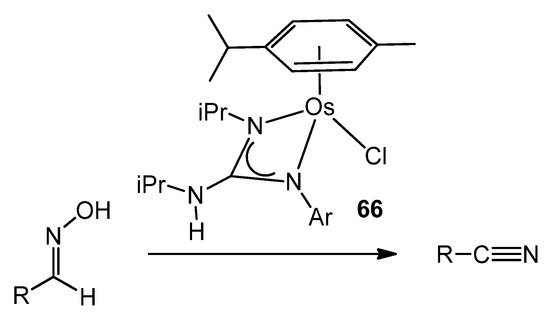
Figure 39. Osmium guanidinato complexes acting as aldoxime dehydration catalysts.
Interestingly, in the search for new coordination modes, guanidines have been modified with other donor groups, such as phosphanes. In this way, ruthenium (II) (Figure 40) and osmium (II) complexes were obtained [137][138].
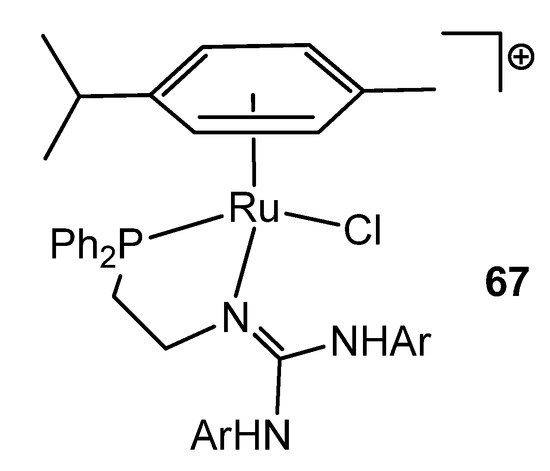
Figure 40. Phosphane-modified guanidine as ligand in a ruthenium complex.
This type of multidentate ligand allows the preparation of the iridium and rhodium guanidinato complexes as 68, which behave as a frustrated Lewis pair (FLP) capable of reversibly activating polar bonds, such as those of water, and non-polar bonds, such as those of dihydrogen (Figure 41) [139][140].
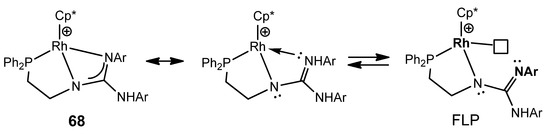
Figure 41. Frustrated Lewis pair based in rhodium phosphanoguanidinato complexes.
Although the usual ligand arrangement in many rhodium complexes is the κ2-N,N chelate [129][141][142], examples are known where the guanidinate acts as a bridge between two metal centers [91], or even a complex where the anionic ligand coordinates the rhodium center in an unusual η5-cyclohexadienyl mode, before isomerizing to the conventional chelate form (Figure 42) [143].
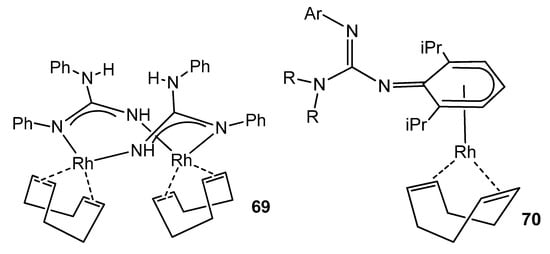
Figure 42. Bridging and arene coordination of guanidinato ligands in rhodium complexes.
Not surprisingly, as well as several examples of rhodium complexes with guanidinato ligands are known, the organometallic chemistry of analogous iridium compounds is wide. In addition to reactivities parallel to those described for rhodium [129][140][142], there are interesting examples of the influence of these ligands on the behavior of the metal. For example, the presence of the lone pair of electrons on the uncoordinated NR2 group increases the electron density of the ligand available for coordination to the metal. This obviously makes these ligands stronger donors than the related amidinato ligands. Consequently, if they coordinate to a metal in a low oxidation state, the possibility of reaching higher oxidation states increase. Thus, in the reactions of iridium guanidinato complexes with O2, the observed reactivity trends correlate well with both the electronic and steric properties of the substituents of the guanidinato ligands, allowing the stabilization and characterization of complexes with peroxo groups (Figure 43) [144][145][146][147].
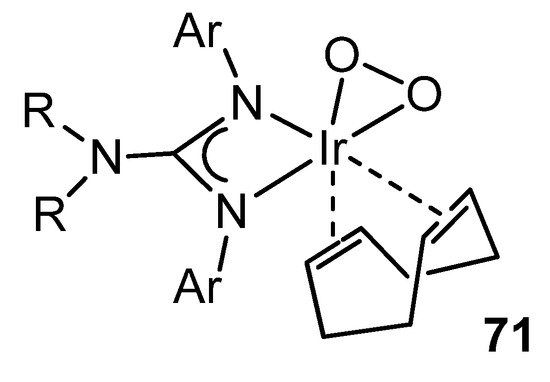
Figure 43. Dioxygen iridium complex.
This same donor capacity of the guanidinato ligand also lies at the basis of several catalytic processes involving organometallic iridium species, which allow the metathesis of carbodiimides and isocyanates, the carbodiimide cleavage or the cross-dimerization of terminal alkynes [148][149][150].
Organometallic iridium (III) complexes are also useful as photophysical active materials in a wide variety of applications, e.g., as lighting devices, as dyes in solar panels, as bio-imaging agents or as photosensitizers for the production of hydrogen from water by photocatalysis [151]. It has been shown that these properties can be altered practically on a whim by modifying the ligands in these complexes. This is why, given the high tunability provided by the guanidinato ligands, they appear as excellent precursors of phosphorescent iridium cyclometalated complexes, becoming prominent in the preparation of Organic Light Emitting Devices (OLEDs), where the proper selection of ligand substituents allows high efficiencies and high variability of the emission color (Figure 44) [152][153][154][155].
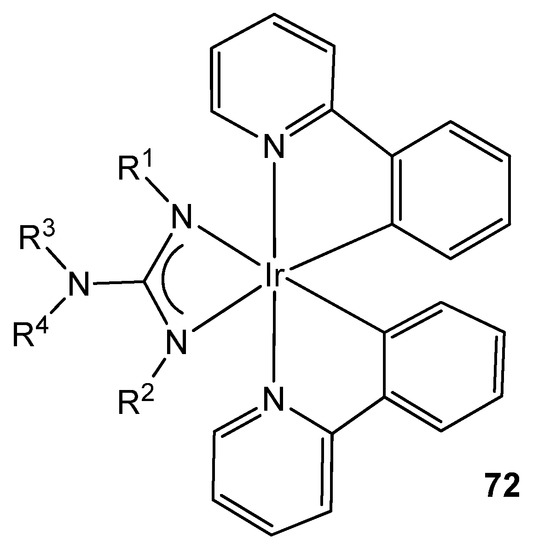
Figure 44. Luminescent iridium guanidinato compound.
The analogy between bulky guanidinates and β-diketiminates has been used to achieve the stabilization of Co(I) toluene adducts, as precursors of dimeric compounds with short Co–Co bonds [156].
As with other platinum group metals, the first reference of group 10 organometallic complexes with guanidinato ligands presents species bearing dianionic ligands with chelate coordination [129][130][131][132]. Since then, it is worth noting the studies carried out by Thirupathi’s group, who have extensively studied aspects of the coordination chemistry of triarylguanidines towards palladium or platinum, in which the presence of Me or Ome substituents on the aromatic rings of the guanidines, together with their inherent basic properties, allowed the formation of interesting six-membered cyclometalated complexes, in which the guanidinato ligand shows a κ2-C,N coordination mode (Figure 45) [157][158][159][160][161][162][163][164][165][166][167].
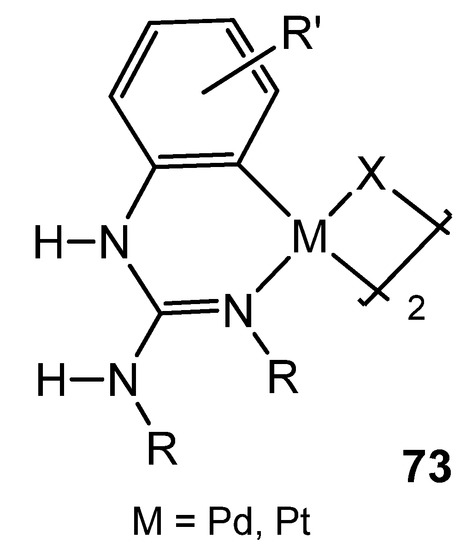
Figure 45. Cyclometalated guanidinato palladium and platinum complexes.
To conclude this section, the organometallic derivatives of group 11 are limited to an example of a strongly phosphorescent copper (I) complex with an (alkyl)(amino)carbene ligand (Figure 46) [168].
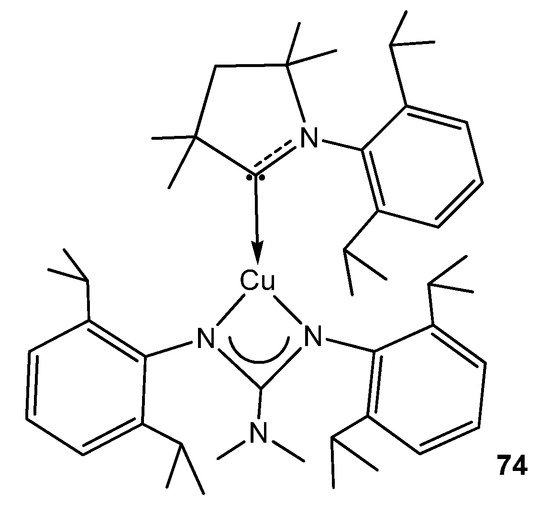
Figure 46. Phosphorescent carbene copper guanidinato compound.
2.3. Group 3 and the Lanthanoid Complexes
The group 3 elements and the lanthanoids represent a family of metals with very interesting properties in terms of chemical reactivity, luminescence or magnetism [169][170]. In particular, the guanidinato derivatives of these metals stand out for their potential use in catalytic processes or as molecular precursors of new ceramic materials. Although there are excellent reviews by Edelmann and Trifonov on rare earth amidinates and guanidinates [171][172][173], here researchers have selected the references focused on organometallic complexes, highlighting their usefulness and the possible differential nuances with the complexes of other groups of metals previously described.
As with some of these examples, the metathesis of a lithium guanidinate and a metal halide is one of the main routes to rare-earth metal derivatives [174][175][176]. This is the case for 75, the first reported organolanthanide complex supported by such a ligand [177]. After the formation of the halide derivative, the reaction with LiCH(SiMe3)2 yields a mononuclear organometallic compound (Figure 47).
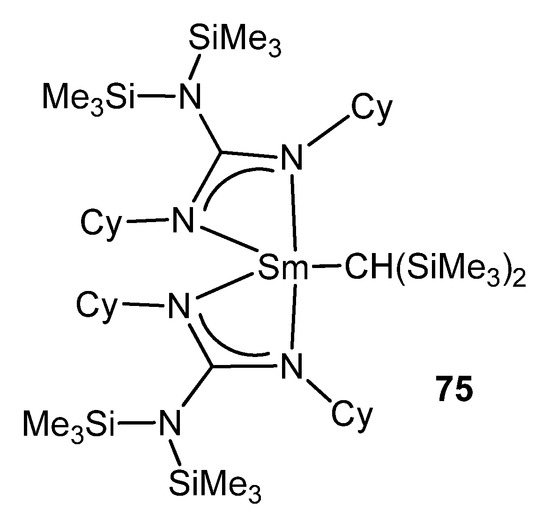
Figure 47. First reported organolanthanide complex with guanidinato ligands.
Stable low-coordinate organometallic complexes, using bulky guanidinato ligands, allow agostic interactions between the yttrium atom and the tert-butyl group in the solid state for [{(Me3Si)2NC(NCy)2}2YtBu] 76 [178] or with two methyl carbon atoms of SiMe3 groups in [{(Me3Si)2NC(NCy)2}2Y(μ-CH2SiMe3)2Li] 77 [179].
Alternatively, the insertion reaction of carbodiimides into M–N bonds of amido derivatives allows a very efficient synthesis of organometallic complexes with symmetric and, mainly, asymmetric guanidinates [180][181].
The presence of NH groups on the ligands assists the insertion and can provide an interesting isomerization of these coordinated fragments (Figure 48) [182][183].
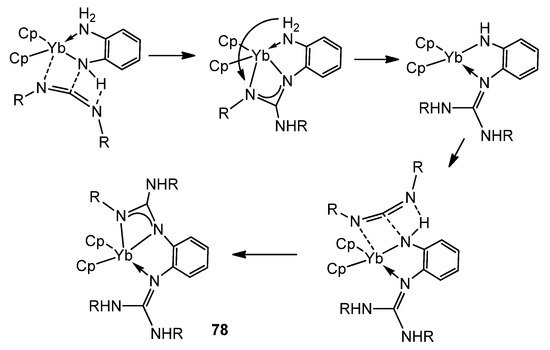
Figure 48. NH-assisted insertion of carbodiimides in amido ytterbium compounds.
In contrast to the usual lack of reactivity observed in guanidinates of other metals, certain organolanthanides allow examples of the insertion of small unsaturated molecules into the metal–guanidinate bond. Complexes [(C5H5)2Ln(μ-η1:η2-N=C(NMe2)2)]2 (Ln = Y 79, Gd 80, Er 81) can mono-insert phenyl isocyanate into the Ln–N bond to yield [(C5H5)2Ln(μ-η1:η2-OC(N=C(NMe2)2)NPh)]2 without further insertion or the usual isocyanate cyclotrimerization [102][184]. The presence of reactive alkyl groups, such as benzyl, offers the possibility to study alternative insertion processes [185][186][187]. Complex 82 inserts nitriles and isocyanates, generating binuclear complexes (Figure 49) [186].
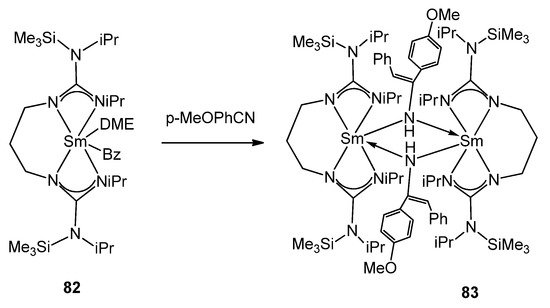
Figure 49. Nitrile insertion in alkyl samarium complex.
A third route is the protonolysis reaction of alkyl derivatives with neutral guanidines [188]. In this way, an alkyl yttrium compound has been obtained with a guanidine with elevated steric demand and with the adequate control of the electronic deficiency of the metal. Compound 84 was highly efficient in the hydrosilylation of alkenes via hydride intermediates with excellent anti-Markovnikov selectivity (Figure 50) [188].
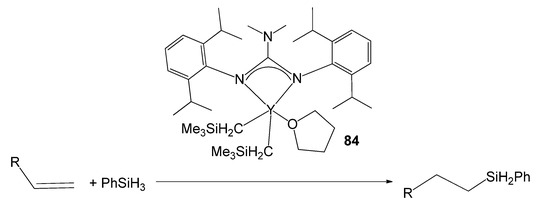
Figure 50. Alkene hydrosilylation with an yttrium guanidinato compound as catalyst.
The catalytic activity of rare-earth metal guanidinates also extends to olefin polymerization processes [189][190][191]. For example, the coordination of two guanidinate ligands allows the stabilization of hydride complexes. The appropriate steric bulk of the substituents on the nitrogen atoms provides high solubility of the lanthanide derivatives and enables Lewis base-free hydrides to be obtained from alkyl derivatives and silanes with an extremely low coordination index on the metal atom. Species such as compound 86 (Figure 51) have shown high catalytic activity in ethylene polymerization [189].
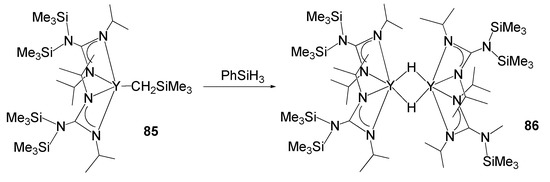
Figure 51. Synthesis of an yttrium hydride stabilized by guanidinato ligands.
Such alkyl or hydride complexes stabilized by guanidinato ligands also allow ring-opening or polar monomer polymerizations [192][193].
To conclude this section, guanidinato ligands have met the needs for rare-earth metal molecular precursors for ALD or CVD techniques: the bidentate coordination mode allows to saturate coordinatively even large cations; the electronic delocalization across the ligand stabilizes the whole molecular structure; and the easy modification of the substituents allows to modulate the physical properties, all contributing to expand the library of potential precursors of new high quality materials obtained by these deposition techniques [194].
3. Conclusions
In conclusion, a great deal of work has been carried out in this field, but there is still scope for the development of new organometallic complexes, in which the choice of metal and the design of the ligands provide greater control of their chemical and physical properties. Guanidines provide easy access to such control, as they can be obtained by very straightforward and efficient syntheses from widely available reagents. The ease of formation of coordination and organometallic complexes, even in unconventional or unstable oxidation states, can be enhanced by potentially modifying these ligands to increase the number and type of donor atoms. A further step towards a firm foothold among the most successful ligands is the possibility to design and synthesize new chiral derivatives for enantioselective catalysis applications. So, again, an appropriate choice of precursors and methods to obtain these ligands, if possible, in a sustainable manner, opens up a whole range of possibilities for organometallic chemists.
References
- Ishikawa, T.; Kumamoto, T. Guanidines in Organic Synthesis. Synthesis 2006, 2006, 737–752.
- Ishikawa, T. Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; John Wiley & Sons: Hoboken, NJ, USA, 2009.
- Berlinck, R.G.S. Natural guanidine derivatives. Nat. Prod. Rep. 1999, 16, 339–365.
- Berlinck, R.G.; Trindade-Silva, A.E.; Santos, M.F. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2012, 29, 1382–1406.
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954.
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. Guanidine Motif in Biologically Active Peptides. Aust. J. Chem. 2014, 67, 965–971.
- Berlinck, R.G.S.; Romminger, S. The chemistry and biology of guanidine natural products. Nat. Prod. Rep. 2016, 33, 456–490.
- Kim, S.-H.; Semenya, D.; Castagnolo, D. Antimicrobial drugs bearing guanidine moieties: A review. Eur. J. Med. Chem. 2021, 216, 113293.
- Oliver, D.W.; Dormehl, I.C.; Wikberg, J.E.S.; Dambrova, M. Guanidines: From molecule to primate. Med. Chem. Res. 2004, 13, 427–438.
- Ishikawa, T. Guanidine Chemistry. Chem. Pharm. Bull. 2010, 58, 1555–1564.
- Fu, X.; Tan, C.-H. Mechanistic considerations of guanidine-catalyzed reactions. Chem. Commun. 2011, 47, 8210–8222.
- Corey, E.J.; Grogan, M. Enantioselective synthesis of alpha-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. J. Org. Lett. 1999, 1, 157–160.
- Ishikawa, T.; Araki, Y.; Kumamoto, T.; Seki, H.; Fukuda, K.; Isobe, T. Modified guanidines as chiral superbases: Application to asymmetric Michael reaction of glycine imine with acrylate or its related compounds. Chem. Commun. 2001, 245–246.
- Shen, J.; Nguyen, T.T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. Chiral Bicyclic Guanidine-Catalyzed Enantioselective Reactions of Anthrones. J. Am. Chem. Soc. 2006, 128, 13692–13693.
- Allingham, M.T.; Howard-Jones, A.; Murphy, P.J.; Thomas, D.A.; Caulkett, P.W.R. Synthesis and applications of C2 Symmetric Guanidine Bases. Tetrahedron Lett. 2003, 44, 8677–8680.
- Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Guanidine-Thiourea Bifunctional Organocatalyst for the Asymmetric Henry (Nitroaldol) Reaction. Adv. Synth. Catal. 2005, 347, 1643–1648.
- Terada, M.; Ube, H.; Yaguchi, Y. Axially Chiral Guanidine as Enantioselective Base Catalyst for 1,4-Addition Reaction of 1,3-Dicarbonyl Compounds with Conjugated Nitroalkenes. J. Am. Chem. Soc. 2006, 128, 1454–1455.
- Fu, X.; Jiang, Z.; Tan, C.-H. Bicyclic guanidine-catalyzed enantioselective phospha-Michael reaction: Synthesis of chiral β-aminophosphine oxides and β-aminophosphines. Chem. Commun. 2007, 5058–5060.
- Ye, W.; Jiang, Z.; Zhao, Y.; Goh, S.L.M.; Leow, D.; Soh, Y.-T.; Tan, C.-H. Chiral Bicyclic Guanidine as a Versatile Brønsted Base Catalyst for the Enantioselective Michael Reactions of Dithiomalonates and β-Keto Thioesters. Adv. Synth. Catal. 2007, 349, 2454–2458.
- Leow, D.; Lin, S.; Chittimalla, S.K.; Fu, X.; Tan, C.-H. Enantioselective Protonation Catalyzed by a Chiral Bicyclic Guanidine Derivative. Angew. Chem. Int. Ed. 2008, 47, 5641–5645.
- Terada, M.; Ikehara, T.; Ube, H. Enantioselective 1, 4-addition reactions of diphenyl phosphite to nitroalkenes catalyzed by an axially chiral guanidine. J. Am. Chem. Soc. 2007, 129, 14112–14113.
- Ye, W.; Xu, J.; Tan, C.T.; Tan, C.-H. 1, 5, 7-Triazabicyclodec-5-ene (TBD) catalyzed Michael reactions. Tetrahedron Lett. 2005, 46, 6875–6878.
- Kita, T.; Georgieva, A.; Hashimoto, Y.; Nakata, T.; Nagasawa, K. C2-Symmetric Chiral Pentacyclic Guanidine: A Phase-Transfer Catalyst for the Asymmetric Alkylation of tert-Butyl Glycinate Schiff Base. Angew. Chem. Int. Ed. 2002, 41, 2832–2834.
- Ishikawa, T.; Isobe, T. Modified guanidines as chiral auxiliaries. Chem. Eur. J. 2002, 8, 553–557.
- Terada, M.; Nakano, M.; Ube, H. Axially chiral guanidine as highly active and enantioselective catalyst for electrophilic amination of unsymmetrically substituted 1, 3-dicarbonyl compounds. J. Am. Chem. Soc. 2006, 128, 16044–16045.
- Pratt, R.C.; Lohmeijer, B.G.G.; Long, D.A.; Waymouth, R.M.; Zhang, S.; He, L.-N. Capture and fixation of CO2 promoted by guanidine derivatives. Aust. J. Chem. 2014, 67, 980–988.
- Mesías-Salazar, Á.; Martínez, J.; Rojas, R.S.; Carrillo-Hermosilla, F.; Ramos, A.; Fernández-Galán, R.; Antiñolo, A. Aromatic guanidines as highly active binary catalytic systems for the fixation of CO2 into cyclic carbonates under mild conditions. Catal. Sci. Technol. 2019, 9, 3879–3886.
- Claver, C.; Yeamin, M.B.; Reguero, M.; Masdeu-Bultó, A.M. Recent advances in the use of catalysts based on natural products for the conversion of CO2 into cyclic carbonates. Green Chem. 2020, 22, 7665–7706.
- Das Neves Gomes, C.; Blondiaux, E.; Thuéry, P.; Cantat, T. Metal-Free Reduction of CO2 with Hydroboranes: Two Efficient Pathways at Play for the Reduction of CO2 to Methanol. Chem. Eur. J. 2014, 20, 7098–7106.
- von Wolff, N.; Lefèvre, G.; Berthet, J.-C.; Thuéry, P.; Cantat, T. Implications of CO2 Activation by Frustrated Lewis Pairs in the Catalytic Hydroboration of CO2: A View Using N/Si+ Frustrated Lewis Pairs. ACS Catal. 2016, 6, 4526–4535.
- Simon, L.; Goodman, J.M. The mechanism of TBD-catalyzed ring-opening polymerization of cyclic esters. J. Org. Chem. 2007, 72, 9656–9662.
- Dong, S.; Feng, X.; Liu, X. Chiral guanidines and their derivatives in asymmetric synthesis. Chem. Soc. Rev. 2018, 47, 8525–8540.
- Katritzky, A.R.; Rogovoy, B.V. Recent developments in guanylating agents. Arkivoc 2005, 4, 49–87.
- Ishikawa, T. Roads to new guanidine chemistry from 2-imidazolidinones through 2-chloroamidinium derivatives. Arkivoc 2006, 7, 148–168.
- Rauws, T.R.M.; Maes, B.U.W. Transition metal-catalyzed N-arylations of amidines and guanidines. Chem. Soc. Rev. 2012, 41, 2463–2497.
- Alonso-Moreno, C.; Antiñolo, A.; Carrillo-Hermosilla, F.; Otero, A. Guanidines: From classical approaches to efficient catalytic syntheses. Chem. Soc. Rev. 2014, 43, 3406–3425.
- Tahir, S.; Badshah, A.; Hussain, R.A. Guanidines from ‘toxic substances’ to compounds with multiple biological applications—Detailed outlook on synthetic procedures employed for the synthesis of guanidines. Bioorg. Chem. 2015, 59, 39–79.
- Chandra, G.; Jenkins, A.D.; Lappert, M.F.; Srivastava, R.C. Amido-derivatives of metals and metalloids. Part X. Reactions of titanium(IV), zirconium(IV), and hafnium(IV) amides with unsaturated substrates, and some related experiments with amides of boron, silicon, germanium, and tin(IV). J. Chem. Soc. A 1970, 2550–2558.
- Jin, G.; Jones, C.; Junk, P.C.; Lippert, K.-A.; Rose, R.P.; Stasch, A. Synthesis and characterisation of bulky guanidines and phosphaguanidines: Precursors for low oxidation state metallacycles. N. J. Chem. 2009, 33, 64–75.
- Barman, M.K.; Baishya, A.; Nembenna, S.J. Bulky guanidinate stabilized homoleptic magnesium, calcium and zinc complexes and their catalytic activity in the Tishchenko reaction. J. Organomet. Chem. 2015, 785, 52–60.
- Maity, A.K.; Fortier, S.; Griego, L.; Metta-Magana, A. Synthesis of a “Super Bulky” Guanidinate Possessing an Expandable Coordination Pocket. J. Inorg. Chem. 2014, 53, 8155–8164.
- Chlupaty, T.; Nevoralova, J.; Ruzickova, Z.; Ruzicka, A. Lithium and Dilithium Guanidinates, a Starter Kit for Metal Complexes Containing Various Mono-and Dianionic Ligands. Inorg. Chem. 2020, 59, 10854–10865.
- Noor, A.; Bauer, T.; Todorova, T.K.; Weber, B.; Gagliardi, L.; Kempe, R. The Ligand-Based Quintuple Bond-Shortening Concept and Some of Its Limitations. Chem. Eur. J. 2013, 19, 9825–9832.
- Bonyhady, S.J.; Green, S.P.; Jones, C.; Nembenna, S.; Stasch, A. A Dimeric Magnesium(I) Compound as a Facile Two-Center/Two-Electron Reductant. Angew. Chem. Int. Ed. 2009, 48, 2973–2977.
- Feil, F.; Harder, S. Guanidinate Complexes of Heavier Alkaline-Earth Metals (Ca, Sr): Syntheses, Structures, Styrene Polymerization and Unexpected Reaction Behaviour. Eur. J. Inorg. Chem. 2005, 4438–4443.
- Cameron, T.M.; Xu, C.; Dipasquale, A.G.; Rheingold, A.L. Synthesis and structure of strontium and barium guanidinates and mixed-ligand guanidinate pentamethylcyclopentadienyl complexes. Organometallics 2008, 27, 1596–1604.
- Moxey, G.J.; Blake, A.J.; Lewis, W.; Kays, D.L. Alkaline Earth Complexes of a Sterically Demanding Guanidinate Ligand. Eur. J. Inorg. Chem. 2015, 5892–5902.
- Sahoo, R.K.; Mahato, M.; Jana, A.; Nembenna, S. Zinc hydride-catalyzed hydrofuntionalization of ketones. J. Org. Chem. 2020, 85, 11200–11210.
- Birch, S.J.; Boss, S.R.; Cole, S.C.; Coles, M.P.; Haigh, R.; Hitchcock, P.B.; Wheatley, A.E.H. The structural characteristics of organozinc complexes incorporating N,N′-bidentate ligands. Dalton Trans. 2004, 3568–3574.
- Khalaf, M.S.; Coles, M.P.; Hitchcock, P.B. A structural, theoretical and coordinative evaluation of the bicyclic guanidinate derived from 1,4,6-triazabicyclooct-4-ene. Dalton Trans. 2008, 4288–4295.
- Zelga, K.; Leszczyński, M.; Justyniak, I.; Kornowicz, A.; Cabaj, M.; Wheatley, A.E.H.; Lewiński, J. Synthesis, structure and unique reactivity of the ethylzinc derivative of a bicyclic guanidine. Dalton Trans. 2012, 41, 5934–5938.
- Coles, M.P.; Hitchcock, P.B. Bicyclic guanidinate compounds of magnesium and their activity as pre-catalysts in the Tishchenko reaction. Eur. J. Inorg. Chem. 2004, 2662–2672.
- Alonso-Moreno, C.; Carrillo-Hermosilla, F.; Garcés, A.; Otero, A.; López-Solera, I.; Rodríguez, A.M.; Antiñolo, A. Simple, versatile, and efficient catalysts for guanylation of amines. Organometallics 2010, 29, 2789–2795.
- Harder, S. Early Main Group Metal. Catalysis: Concepts and Rections; Wiley-VCH Verlag GmbH & Co. KGaAL: Weinheim, Germany, 2020; Volume 12, p. 69469.
- Aeilts, S.L.; Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum alkyl complexes containing guanidinate ligands. Organometallics 1998, 17, 3265–3270.
- Koller, J.; Bergman, R.G. Synthesis, characterization, and reactivity of aluminum alkyl/amide complexes supported by guanidinate and monoanionic OCO-pincer ligands. Organometallic 2010, 29, 3350–3356.
- Koller, J.; Bergman, R.G. Highly efficient aluminum-catalyzed hydro-amination/-hydrazination of carbodiimides. Organometallics 2010, 29, 5946–5952.
- Cole, M.L.; Davies, A.J.; Jones, C.; Junk, P.C.; McKay, A.I.; Stasch, A.Z. Aluminum and indium complexes derived from guanidines, triazenes, and amidines. Anorg. Allg. Chem. 2015, 641, 2233–2244.
- Han, H.-F.; Zhang, S.-F.; Guo, Z.-Q.; Tong, H.-B.; Wei, X.-H. Three asymmetric guanidinato metal complexes: Synthesis, crystal structures and their use as pre-catalysts in the Meerwein–Ponndorf–Verley reduction. Polyhedron 2015, 99, 71–76.
- Chang, C.-C.; Hsiung, C.-S.; Su, H.-L.; Srinivas, B.; Chiang, M.Y.; Lee, G.-H.; Wang, Y. Carbodiimide insertion into organoaluminum compounds and thermal rearrangement of the products. Organometallics 1998, 17, 1595–1601.
- Zhang, W.-X.; Li, D.; Wang, Z.; Xi, Z. Alkyl aluminum-catalyzed addition of amines to carbodiimides: A highly efficient route to substituted guanidines. Organometallics 2009, 28, 882–887.
- Wei, Y.; Wang, S.; Zhou, S.; Feng, Z.; Guo, L.; Zhu, X.; Mu, X.; Yao, F. Aluminum Alkyl Complexes Supported by Bidentate N,N Ligands: Synthesis, Structure, and Catalytic Activity for Guanylation of Amines. Organometallics 2015, 34, 1882–1889.
- Riddlestone, I.M.; Urbano, J.; Phillips, N.; Kelly, M.J.; Vidovic, D.; Bates, J.I.; Taylor, R.; Aldridge, S. Salt metathesis for the synthesis of M–Al and M–H–Al bonds. Dalton Trans. 2013, 42, 249–258.
- Bayram, M.; Bläser, D.; Wölper, C.; Schulz, S. Syntheses and Structures of Bis-Amidinate–Alane Complexes. Organometallics 2014, 33, 2080–2087.
- Chlupatý, T.; Bílek, M.; Moncol’, J.; Ruzicková, Z.; Ruzicka, A. Addition of dimethylaluminium chloride to N,N′-Disubstituted carbodiimides. J. Organomet. Chem. 2015, 786, 48–54.
- Han, H.-F.; Guo, Z.-Q.; Zhang, S.-F.; Li, J.; Wei, X.-H. Guanidinato aluminum complexes: Synthesis, crystal structures and reactivities. RSC Adv. 2016, 6, 101437–101446.
- Han, H.; Guo, Z.; Zhang, S.; Hua, Y.; Wei, X. Synthesis and crystal structures of guanidinatoaluminum complexes and catalytic study for MPV reduction. Polyhedron 2017, 126, 214–219.
- Peddarao, T.; Sarkar, N.; Nembenna, S. Mono- and Bimetallic Aluminum Alkyl, Alkoxide, Halide and Hydride Complexes of a Bulky Conjugated Bis-Guanidinate (CBG) Ligand and Aluminum Alkyls as Precatalysts for Carbonyl Hydroboration. Inorg. Chem. 2020, 59, 4693–4702.
- Sarkar, N.; Sahoo, R.K.; Mukhopadhyay, S.; Nembenna, S. Organoaluminum Cation Catalyzed Selective Hydrosilylation of Carbonyls, Alkenes, and Alkynes. Eur. J. Inorg. Chem. 2022, e202101030.
- Rösch, A.; Seifert, F.; Vass, V.; Görls, H.; Kretschmer, R. Synthesis, structure, and catalytic activity of dinuclear aluminium bis (amidinate) and bis (guanidinate) complexes. New J. Chem. 2021, 45, 972–981.
- Rios Yepes, Y.; Mesías-Salazar, Á.; Becerra, A.; Daniliuc, C.G.; Ramos, A.; Fernández-Galán, R.; Rodríguez-Diéguez, A.; Antiñolo, A.; Carrillo-Hermosilla, F.; Rojas, R.S. Mono- and Dinuclear Asymmetric Aluminum Guanidinates for the Catalytic CO2 Fixation into Cyclic Carbonates. Organometallics 2021, 40, 2859–2869.
- Fernández-Galán, R.; Ramos, A.; Huergo, E.; Antiñolo, A.; Carrillo-Hermosilla, F.; Rodríguez-Diéguez, A.; García-Vivó, D. Unusual ligand rearrangement: From N-phosphinoguanidinato to phosphinimine-amidinato compounds. Chem. Commun. 2019, 55, 2809–2812.
- Huergo, E.; Fernández-Galán, R.; Ramos, A.; Antiñolo, A.; Carrillo-Hermosilla, F.; Rodríguez-Diéguez, A.; García-Vivó, D. Reactivity of N-Phosphinoguanidines of the Formula (HNR)(Ph2PNR)C(NAr) toward Main Group MetalAlkyls: Facile Ligand Rearrangement from N-Phosphinoguanidinates to Phosphinimine-Amidinates. Inorg. Chem. 2020, 59, 15262–15275.
- Brazeau, A.L.; Barry, S.T. Atomic layer deposition of aluminum oxide thin films from a heteroleptic, amidinate-containing precursor. Chem. Mat. 2008, 20, 7287–7291.
- Dodonov, V.A.; Xiao, L.; Kushnerova, O.A.; Baranov, E.V.; Zhao, Y.; Yang, X.J.; Fedushkin, I.L. Transformation of carbodiimides to guanidine derivatives facilitated by gallylenes. Chem Commun. 2020, 56, 7475–7478.
- Horeglad, P.; Litwińska, A.; Zukowska, G.Z.; Kubicki, D.; Szczepaniak, G.; Dranka, M.; Zachara, J. The influence of organosuperbases on the structure and activity of dialkylgallium alkoxides in the polymerization of rac -lactide: The road to stereo diblock PLA copolymers. App. Organomet. Chem. 2013, 27, 328–336.
- Kassymbek, A.; Britten, J.F.; Spasyuk, D.; Gabidullin, B.; Nikonov, G.I. Interaction of multiple bonds with NacNacGa: Oxidative cleavage vs coupling and cyclization. Inorg. Chem. 2019, 58, 8665–8672.
- Jin, G.; Jones, C.; Junk, P.C.; Stasch, A.; Woodul, W.D. Group 13 metal (I) and (II) guanidinate complexes: Effect of ligand backbone on metal oxidation state and coordination sphere. New J. Chem. 2008, 32, 835–842.
- Jones, C.; Junk, P.C.; Platts, J.A.; Stasch, A. Four-membered group 13 metal(I) N-heterocyclic carbene analogues: Synthesis, characterization, and theoretical studies. J. Am. Chem. Soc. 2006, 128, 2206–2207.
- Matsuda, I.; Itoh, K.; Ishii, Y. Reactions of CO2 and CO2 Analogs (CXY with X, Y = O, S, NR) with Reagents Containing Si–H and Si–N Units. J. Organomet. Chem. 1974, 69, 353–359.
- Das, S.; Pati, S.K. Computational Exploration of Intramolecular Sn/N Frustrated Lewis Pairs for Hydrogen Activation and Catalytic Hydrogenation. Organometallics 2021, 40, 194–202.
- George, T.A.; Jones, K.; Lappert, M.F. Amino-derivatives of metals and metalloids. Part II. Ainostannylation of unsaturated substrates, and the infrared spectra and structures of carbamato- and dithiocarbamato-trimethylstannanes and related compounds. J. Chem. Soc. 1965, 2157–2165.
- Hänssgen, D.; Odenhausen, E. Organotin heterocycles. J. Organomet. Chem. 1977, 124, 143–150.
- Hänssgen, D.; Pohl, I. Ringerweiterungs-und-spaltungsreaktionen der Cyclodistannazane. Chem. Ber. 1979, 112, 2798–2803.
- Kupchik, E.J.; Hanke, H.E.; DiMarco, J.P.; Chessari, R.J. Synthesis of N,N,N′-trisubstituted N′′-cyanoguanidines and N-aryl-N′-(triorganostannyl)-N′N′′-dicyanoguanidines. J. Chem. Eng. Data 1981, 26, 105–106.
- Andrade-López, N.; Ariza-Castolo, A.; Contreras, R.; Vázquez-Olmos, A.; Barba-Behrens, N.; Tlahuext, H. Versatile behavior of 2-guanidinobenzimidazole nitrogen atoms toward protonation, coordination and methylation. Heteroat. Chem. 1997, 8, 397–410.
- Fialon, M.-P.; Andrade-Lopez, N.; Barba-Behrens, N.; Contreras, R. Organometallic tin complexes derived from 2-guanidinobenzimidazole. Heteroat. Chem. 1998, 9, 637–641.
- Wood, D.; Yap, G.P.A.; Richeson, D.S. N-Substituted Guanidinate Anions as Ancillary Ligands in Group 4 Chemistry. Syntheses and Characterization of M2Cl2, − (M = Zr, Hf; R = iPr, Cy), and Zr(CH2Ph)3. Inorg. Chem. 1999, 38, 5788–5794.
- Giesbrecht, G.R.; Whitener, G.D.; Arnold, J. Crystal Packing Forces Dictate η1- versus η2-Coordination of Benzyl Groups in Zr(CH2Ph)3. Organometallics 2000, 19, 2809–2812.
- Coles, M.P.; Hitchcock, P.B. Exploration of the suitability of bicyclic guanidinates as ligands in catalytic chemistry mediated by titanium. Organometallics 2003, 22, 5201–5211.
- Fandos, R.; Otero, A.; Rodríguez, A.; Terreros, P. Syntheses of new titanium(IV) and rhodium(I) guanidinido complexes. Collect. Czech. Chem. Commun. 2007, 72, 579–588.
- Rodriguez, G.; Sperry, C.K.; Bazan, G.C. Boratabenzene complexes of zirconium, hafnium and chromium. J. Mol. Catal. A 1998, 128, 5–28.
- Fernández-Galán, R.; Antiñolo, A.; Carrillo-Hermosilla, F.; López-Solera, I.; Otero, A.; Serrano-Laguna, A.; Villaseñor, E. New zirconium and zirconocene guanidinate complexes. J. Organomet. Chem. 2012, 711, s35–s42.
- Zuckerman, R.L.; Bergman, R.G. Structural factors that influence the course of overall cycloaddition reactions between imidozirconocene complexes and heterocumulenes. Organometallics 2000, 19, 4795–4809.
- Zuckerman, R.L.; Bergman, R.G. Mechanistic investigation of cycloreversion/cycloaddition reactions between zirconocene metallacycle complexes and unsaturated organic substrates. Organometallics 2001, 20, 1792–1807.
- Duncan, A.P.; Mullins, S.M.; Arnold, J.; Bergman, R.G. Synthesis, structural investigation, and reactivity of neutral and cationic bis (guanidinato) zirconium(IV) complexes. Organometallics 2001, 20, 1808–1819.
- Bazinet, P.; Wood, D.; Yap, G.P.A.; Richeson, D.S. Synthesis and Structural Investigation of N,N′,N′′-Trialkylguanidinato-Supported Zirconium(IV) Complexes. Inorg. Chem. 2003, 42, 6225–6229.
- Ong, T.-G.; Yap, G.P.A.; Richeson, D.S. Redefining the Coordination Geometry and Reactivity of Guanidinate Complexes by Covalently Linking the Guanidinate Ligands. Synthesis and Reactivity of M(CH2Ph)2 (R = iPr; M = Ti, Zr). Organometallics 2003, 22, 387–389.
- Ong, T.-G.; Yap, G.P.A.; Richeson, D.S. Formation of a guanidinate-supported titanium imido complex: A catalyst for alkyne hydroamination. Organometallics 2002, 21, 2839–2841.
- Ong, T.-G.; Wood, D.; Yap, G.P.A.; Richeson, D.S. Transformations of aryl isocyanide on guanidinate-supported organozirconium complexes to yield terminal imido, iminoacyl, and enediamido ligands. Organometallics 2002, 21, 1–3.
- Fernández-Galán, R.; Antiñolo, A.; Carrillo-Hermosilla, F.; López-Solera, I.; Otero, A.; Serrano-Laguna, A.; Villaseñor, E. Migratory Insertion Reactions in Asymmetrical Guanidinate-Supported Zirconium Complexes. Organometallics 2012, 31, 8360–8369.
- Zhang, J.; Han, F.; Han, Y.; Chen, Z.; Zhou, X. Synthesis and structures of titanium and yttrium complexes with N,N′-tetramethylguanidinate ligands: Different reactivity of the M–N bonds toward phenyl isocyanate. Dalton Trans. 2009, 1806–1811.
- Zhou, M.; Yang, Q.; Tong, H.; Yan, L.; Wang, X. Organoamido zirconium(IV) and titanium(IV) complexes and their catalysis towards ethylene polymerization. RSC Adv. 2015, 5, 105292–105298.
- Shen, H.; Chan, H.-S.; Xie, Z. Guanylation of amines catalyzed by a half-sandwich titanacarborane amide complex. Organometallics 2006, 25, 5515–5517.
- Shen, H.; Chan, H.-S.; Xie, Z. Reaction of Zr(NMe2)(DME) with Guanidines: Metallacarborane-Mediated C−N Bond Cleavage and 1,5-Sigmatropic Rearrangement. J. Am. Chem. Soc. 2007, 129, 12934–12935.
- Gott, A.L.; Coles, S.R.; Clarke, A.J.; Clarkson, G.J.; Scott, P. Chiral alkoxide-functionalized guanidinates from ring-opening rearrangement of aminooxazolinate complexes. Organometallics 2007, 26, 136–142.
- Gott, A.L.G.; Clarkson, J.; Deeth, R.J.; Hammond, M.L.; Morton, C.; Scott, P. Constrained geometry aminooxazolinate ligands giving chiral zirconium guanidinates; catalytic cyclohydroamination. Dalton Trans. 2008, 2983–2990.
- Potts, S.E.; Carmalt, C.J.; Blackman, C.S.; Abou-Chahine, F.; Pugh, D.; Davies, H.O. Synthesis of zirconium guanidinate complexes and the formation of zirconium carbonitride via low pressure CVD. Organometallics 2009, 28, 1838–1844.
- Wasslen, Y.A.; Tois, E.; Haukka, S.; Kreisel, K.A.; Yap, G.P.A.; Halls, M.D.; Barry, S.T. A family of heteroleptic titanium guanidinates: Synthesis, thermolysis, and surface reactivity. Inorg. Chem. 2010, 49, 1976–1982.
- Xu, K.; Milanov, A.P.; Winter, M.; Barreca, D.; Gasparotto, A.; Becker, H.-W.; Devi, A. Heteroleptic Guanidinate- and Amidinate-Based Complexes of Hafnium as New Precursors for MOCVD of HfO2. Eur. J. Inorg. Chem. 2010, 1679–1688.
- Kurek, A.; Gordon, P.G.; Karle, S.; Devi, A.; Barry, S.T. Recent advances using guanidinate ligands for chemical vapour deposition (CVD) and atomic layer deposition (ALD) applications. Aust. J. Chem. 2014, 67, 989–996.
- Hirotsu, M.; Fontaine, P.P.; Zavalij, P.Y.; Sita, L.R. Extreme N⋮N Bond Elongation and Facile N-Atom Functionalization Reactions within Two Structurally Versatile New Families of Group 4 Bimetallic “Side-on-Bridged” Dinitrogen Complexes for Zirconium and Hafnium. J. Am. Chem. Soc. 2007, 129, 12690–12692.
- Aguilar-Calderón, J.R.; Metta-Magaña, A.J.; Noll, B.; Fortier, S. Reversible oxidative-addition and reductive-elimination of thiophene from a titanium complex and its thermally-induced hydrodesulphurization chemistry. Angew. Chem. Int. Ed. 2016, 55, 1–6.
- Giménez-Torres, A.; Aguilar-Calderón, J.R.; Encerrado-Manríquez, A.M.; Pink, M.; Metta-Magaça, A.J.; Lee, W.-Y.; Fortier, S. Titanium-Mediated Catalytic Hydrogenation of Monocyclic and Polycyclic Arenes. Chem. Eur. J. 2020, 26, 2803–2807.
- Aguilar-Calderón, J.R.; Murillo, J.; Gómez-Torres, A.; Saucedo, C.; Jordan, A.; Metta-Magaña, A.J.; Pink, M.; Fortier, S. Redox character and small molecule reactivity of a masked titanium(II) synthon. Organometallics 2020, 39, 295–311.
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; López-Solera, I.; Menot, B.; Fernández-Galán, R.; Villaseñor, E.; Otero, A. New alkylimido niobium complexes supported by guanidinate ligands: Synthesis, characterization, and migratory insertion reactions. Organometallics 2012, 31, 1840–1848.
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; López-Solera, I.; Fernández-Galán, R.; Serrano, A.; Villaseñor, E. Synthesis, characterization and reactivity of new dinuclear guanidinate diimidoniobium complexes. Eur. J. Inorg. Chem. 2013, 2940–2946.
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; Suárez, F.J.; López-Solera, I.; Fernández-Galán, R.; Villaseñor, E. Asymmetric niobium guanidinates as intermediates in the catalytic guanylation of amines. Dalton Trans. 2013, 42, 8223–8230.
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; López-Solera, I.; Fernández-Galán, R.; Villaseñor, E. Unexpected mild C–N bond cleavage mediated by guanidine coordination to a niobium iminocarbamoyl complex. Chem. Commun. 2013, 49, 8701–8703.
- Thirupathi, N.; Yap, G.P.A.; Richeson, D.S. Mono- and Dianionic Guanidinate Ligands. Reactivity of Ta(NMe2)3 and TaCl(NMe2)3 with Me3SiCl and ArNC (Ar = 2,6-Me2C6H4). Organometallics 2000, 19, 2573–2579.
- Mohammad, A.; Olson, J.R.; Rotsch, D.A.; Bemowski, R.D.; Swenson, D.C.; Messerle, L. High-and Mid-Valent Tantalum and Mono (peralkylcyclopentadienyl) tantalum Complexes of the Bicyclic Guanidinate Hexahydropyrimidopyrimidinate. Organometallics 2013, 32, 6232–6239.
- Yonke, B.L.; Keane, A.J.; Zavalij, P.Y.; Sita, L.R. Mononuclear Tantalum(IV, d1) Imido Complexes Supported by the Monocyclopentadienyl, Amidinate and Guanidinate Ligand Sets As Models to Explore Dinitrogen Fixation by “End-On-Bridged” Dinuclear {}2(μ-η1:η1-N2) Complexes. Organometallics 2012, 31, 345–355.
- Keane, A.J.; Yonke, B.L.; Hirotsu, M.; Zavalij, P.Y.; Sita, L.R. Fine-Tuning the Energy Barrier for Metal-Mediated Dinitrogen N≡N Bond Cleavage. J. Am. Chem. Soc. 2014, 136, 9906–9909.
- Saadati, F.; Griffin, S.E.; Schafer, L.L. Guanidinate Early-Transition-Metal Complexes: Efficient and Selective Hydroaminoalkylation of Alkenes. Organometallics 2022, 41, 1816–1822.
- Hao, H.; Cui, C.; Bai, G.; Roesky, H.W.; Noltemeyer, M.; Schmidt, H.-G.; Ding, Y.Z. Bis(arylimido) Molybdenum(VI) Amidinate and Guanidinate Complexes; Molecular Structures of (Ar = 2,6-i -Pr2C6H3; Cy = Cyclohexyl) and · . Anorg. Allg. Chem. 2000, 626, 1660–1664.
- Wang, L.; Hu, L.; Zhang, H.; Chen, H.; Deng, L. Three-coordinate iron(IV) bisimido complexes with aminocarbene ligation: Synthesis, structure, and reactivity. J. Am. Chem. Soc. 2015, 137, 14196–14207.
- Gao, Y.; Carta, V.; Pink, M.; Smith, J.M. Catalytic Carbodiimide Guanylation by a Nucleophilic, High Spin Iron(II) Imido Complex. J. Am. Chem. Soc. 2021, 143, 5324–5329.
- Bailey, P.J.; Mitchell, L.A.; Parsons, S. Guanidine anions as chelating ligands; syntheses and crystal structures of and . J. Chem. Soc. Dalton Trans. 1996, 2839–2841.
- Dinger, M.B.; Henderson, W.; Nicholson, B.K. Organometallic complexes of platinum-group metals incorporating substituted guanidine dianion (triazatrimethylenemethane) ligands. J. Organomet. Chem. 1998, 556, 75–88.
- Singh, T.; Kishan, R.; Nethaji, M.; Thirupathi, N. Synthesis, Reactivity Studies, Structural Aspects, and Solution Behavior of Half Sandwich Ruthenium(II) N,N′,N″-Triarylguanidinate Complexes. Inorg. Chem. 2012, 51, 157–169.
- Kishan, R.; Kumar, R.; Baskaran, S.; Sivasankar, C.; Thirupathi, N. Ionic and Neutral Half-Sandwich Guanidinatoruthenium(II) Complexes and Their Solution Behavior. Eur. J. Inorg. Chem. 2015, 3182–3194.
- Bailey, P.J.; Grant, K.J.; Mitchell, L.A.; Pace, S.; Parkin, A.; Parsons, S. Guanidinates as chelating anionic ligands for early, middle and late transition metals: Syntheses and crystal structures of , and . J. Chem. Soc. Dalton Trans. 2000, 1887–1891.
- Robin Kumar, R.; Ujjval, R.; Thirupathi, N. Half Sandwich Electron Deficient N,N′,N′′-Triarylguanidinato-ruthenium(II) Complexes: Syntheses, Reactivity Studies, and Structural Aspects. Eur. J. Inorg. Chem. 2019, 31, 3619–3628.
- García-Álvarez, R.; Suárez, F.J.; Dıéz, J.; Crochet, P.; Cadierno, V.; Antiñolo AFernández-Galán, R.; Carrillo-Hermosilla, F. Ruthenium(II) Arene Complexes with Asymmetrical Guanidinate Ligands: Synthesis, Characterization, and Application in the Base-Free Catalytic Isomerization of Allylic Alcohols. Organometallics 2012, 31, 8301–8311.
- Menéndez-Rodríguez, L.; Tomás-Mendivil, E.; Francos, J.; Crochet, P.; Cadierno, V.; Antiñolo, A.; Fernández-Galán, R.; Carrillo-Hermosilla, F. Reactivity of the Dimer (C10H16 = 2,7-Dimethylocta-2,6-diene-1,8-diyl) toward Guanidines: Access to Ruthenium(IV) and Ruthenium(II) Guanidinate Complexes. Organometallics 2015, 34, 2796–2809.
- Francos, J.; Gonzalez-Liste, P.J.; Menéndez-Rodríguez, L.; Crochet, P.; Cadierno, V.; Borge, J.; Antinolo, A.; Fernández-Galán, R.; Carrillo-Hermosilla, F. Half-Sandwich Guanidinate–Osmium(II) Complexes: Synthesis and Application in the Selective Dehydration of Aldoximes. Eur. J. Inorg. Chem. 2016, 393–402.
- Wilkinson, E.-T.; Viguri, F.; Rodríguez, R.; López, J.A.; García-Orduña, P.; Lahoz, F.J.; Lamata, P.; Carmona, D. Strained Ruthenium Complexes Bearing Tridentate Guanidine-Derived Ligands. Helv. Chim. Acta 2021, 104, e2100044.
- Parker, A.; Lamata, P.; Viguri, F.; Rodríguez, R.; López, J.A.; Lahoz, F.J.; García-Orduña, P.; Carmona, D. Half-sandwich complexes of osmium containing guanidine-derived ligands. Dalton Trans. 2020, 49, 13601–13617.
- Carmona, M.; Ferrer, J.; Rodríguez, R.; Passarelli, V.; Lahoz, F.J.; García-Orduña, P.; Cañadillas-Delgado, L.; Carmona, D. Reversible Activation of Water by an Air- and Moisture-Stable Frustrated Rhodium Nitrogen Lewis Pair. Chem. Eur. J. 2019, 25, 13665–13670.
- Ferrer, C.; Ferrer, J.; Passarelli, V.; Lahoz, F.J.; García-Orduña, P.; Carmona, D. Well-Stabilized but Strained Frustrated Lewis Pairs Based on Rh/N and Ir/N Couples. Organometallics 2022, 41, 1445–1453.
- Kumar, R.; Kishan, R.; Thomas, J.M.; Chinnappan, S.; Thirupathi, N. Probing the factors that influence the conformation of a guanidinato ligand in (NN = chelating N,N′,N′′-tri(o-substituted aryl)guanidinate(1−); X = chloro, azido and triazolato). New J. Chem. 2018, 42, 1853–1866.
- Kumar, R.; Thirupathi, N. Syntheses, characterisation, and catalytic role of (η⁵-C₅Me₅)Rh(III) guanidinato complexes in transfer hydrogenation (TH) and TH–etherification. RSC Adv. 2017, 7, 33890–33904.
- Jones, C.; Mills, D.P.; Stasch, A. Flexible coordination of bulky amidinates and guanidinates towards rhodium(I): Conversion of kinetic to thermodymanic isomers. Dalton Trans. 2008, 4799–4804.
- Rohde, J.-U.; Kelley, M.R.; Lee, W.-T. Synthesis, Characterization, and O2 Reactivity of Iridium(I) Complexes Supported by Guanidinato Ligands. Inorg. Chem. 2008, 47, 11461–11463.
- Rohde, J.-U.; Lee, W.-T. Stabilization of Iridium(IV) by Monoanionic Dialkyldiarylguanidinato Ligands. J. Am. Chem. Soc. 2009, 131, 9162–9163.
- Kelley, M.R.; Rohde, J.-U. Evidence for a reactive (alkene)peroxoiridium(III) intermediate in the oxidation of an alkene complex with O2. Chem. Commun. 2012, 48, 2876–2878.
- Kelley, M.R.; Rohde, J.-U. Guanidinato Complexes of Iridium: Ligand-Donor Strength, O2 Reactivity, and (Alkene)peroxoiridium(III) Intermediates. Inorg. Chem. 2013, 52, 2564–2580.
- Holland, A.W.; Bergman, R.G. Heterocumulene Metathesis by Iridium Guanidinate and Ureylene Complexes: Catalysis Involving Reversible Insertion to Form Six-Membered Metallacycles. J. Am. Chem. Soc. 2002, 124, 9010–9011.
- Holland, A.W.; Bergman, R.G. Thermal reactivity of Group 9 transition metal pinacolate complexes: Heterocumulene cleavage and CO bond formation. Inorg. Chim. Acta 2002, 341, 99–106.
- Ogata, K.; Oka, O.; Toyota, A.; Suzuki, N.; Fukuzawa, S. Phosphine-Dependent Selective Cross-Dimerization between Terminal Alkylacetylene and Silylacetylene by Iridium(I) Guanidinate Complex-Phosphine System. Synlett 2008, 2663–2666.
- Xu, H.; Chen, R.; Sun, Q.; Lai, W.; Su, Q.; Huang, W.; Liu, X. Recent progress in metal–organic complexes for optoelectronic applications. Chem. Soc. Rev. 2014, 43, 3259–3302.
- Rai, V.K.; Nishiura, M.; Takimoto, M.; Zhao, S.; Liu, Y.; Hou, Z. Bis-Cyclometalated Iridium(III) Complexes Bearing Ancillary Guanidinate Ligands. Synthesis, Structure, and Highly Efficient Electroluminescence. Inorg. Chem. 2012, 51, 822–835.
- Feng, Y.; Li, P.; Zhuang, X.; Ye, K.; Peng, T.; Liu, Y.; Wang, Y. A novel bipolar phosphorescent host for highly efficient deep-red OLEDs at a wide luminance range of 1000–10000 cdm−2. Chem. Commun. 2015, 51, 12544–12547.
- Hasan, K.; Pal, A.K.; Auvray, T.; Zysman-Colman, E.; Hanan, G.S. Blue-green emissive cationic iridium(III) complexes using partially saturated strongly-donating guanidyl-pyridine/-pyrazine ancillary ligands. Chem. Commun. 2015, 51, 14060–14063.
- Kabir, E.; Wu, Y.; Sittel, S.; Nguyen, B.-L.; Teets, T.S. Improved deep-red phosphorescence in cyclometalated iridium complexes via ancillary ligand modification. Inorg. Chem. Front. 2020, 7, 1362–1373.
- Jones, C.; Schulten, C.; Rose, R.P.; Stasch, A.; Aldridge, S.; Woodul, W.D.; Murray, K.S.; Moubaraki, B.; Brynda, M.; La Macchia, G.; et al. Amidinato– and Guanidinato–Cobalt(I) Complexes: Characterization of Exceptionally Short Co–Co Interactions. Angew. Chem. Int. Ed. 2009, 48, 7406–7410.
- Gopi, K.; Thirupathi, N. Synthesis, Reactivity, Structural Aspects, and Solution Dynamics of Cyclopalladated Compounds of N,N′,N′′-Tris(2-anisyl)guanidine. Organometallics 2011, 30, 572–583.
- Elumalai, P.; Thirupathi, N.; Nethaj, M. Six-membered cyclopalladated sym N,N′,N″-tri(4-tolyl)guanidines: Synthesis, reactivity studies and structural aspects. J. Organomet. Chem. 2013, 741–742, 141–147.
- Gopi, K.; Saxena, P.; Nethaji, M.; Thirupathi, N. Influence of steric effect on the structural aspects of N,N′,N″-triarylguanidine derived six-membered palladacycles. Polyhedron 2013, 52, 1041–1052.
- Elumalai, P.; Thirupathi, N.; Nethaji, M. Dual Role of Acetate as a Nucleophile and as an Internal Base in Cycloplatination Reaction of sym-N,N′,N″-Triarylguanidines. Inorg. Chem. 2013, 52, 1883–1894.
- Mishra, V.; Thirupathi, N. Critical Role of Anions in Platinum(II) Precursors upon the Structural Motifs of Six-Membered Cycloplatinated N,N′,N″-Triarylguanidines. ACS Omega 2018, 3, 6075–6090.
- Ujjval, R.; Deepa, M.; Thomas, J.M.; Sivasankar, C.; Thirupathi, N. Unusual + → + Coordination Mode Flip of the Guanidinate(1−) Ligand in Cationic N,N′,N″-Tris(3,5-xylyl)guanidinatoplatinum(II) Bis(phosphine) Complexes. Syntheses, Structural and Theoretical Studies. Organometallics 2020, 39, 3663–3678.
- Thakur, V.; Thirupathi, N. Syntheses and structural aspects of dinuclear cycloplatinated N,N′,N″-triarylguanidinate(2−) complexes with a novel tridentate μ2-κ2(C,N):κ1N coordination mode. J. Organomet. Chem. 2020, 911, 121138–121143.
- Mishra, V.; Sinha, N.K.; Thirupathi, N. Reactions of Cycloplatinated Guanidine Complexes with Hg(OC(O)CF3)2: Formation of a One-Dimensional Coordination Polymer Containing a Pt2Hg(μ2-S(O)Me2-S,O) Repeating Unit versus a Discrete Pt2Hg2 Complex. Inorg. Chem. 2021, 60, 3879–3892.
- Sinha, N.K.; Thirupathi, N. CNN Palladium(II) Pincer Complexes Containing N-Substituted Monoanionic and Dianionic Guanidinate Ligands: Syntheses, Structural Aspects, and Their Utility in Suzuki–Miyaura Coupling Reactions. Organometallics 2021, 40, 3535–3549.
- Thakur, V.; Thirupathi, N. Reactivity studies of cycloplatinated triarylguanidinato complexes, (X = Cl and OC(O)CF3) towards Lewis bases, and structural aspects of the products. J. Organomet. Chem. 2022, 959, 122200–122210.
- Saxena, P.; Thomas, J.M.; Sivasankarb, C.; Thirupathi, N. Syntheses and structural aspects of six-membered palladacyclic complexes derived from N,N′,N′′-triarylguanidines with N- or S-thiocyanate ligands. New J. Chem. 2019, 43, 2307–2327.
- Romanov, A.S.; Chotard, F.; Rashid, J.; Bochmann, M. Synthesis of copper(I) cyclic (alkyl)(amino)carbene complexes with potentially bidentate N^N, N^S and S^S ligands for efficient white photoluminescence. Dalton Trans. 2019, 48, 15445–15454.
- Day, B.M.; Guo, F.-S.; Layfield, R.A. Cyclopentadienyl Ligands in Lanthanide Single-Molecule Magnets: One Ring to Rule Them All? Acc. Chem. Res. 2018, 51, 1880–1889.
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148.
- Edelmann, F.T. Lanthanide amidinates and guanidinates: From laboratory curiosities to efficient homogeneous catalysts and precursors for rare-earth oxide thin films. Chem. Soc. Rev. 2009, 38, 2253–2268.
- Edelmann, F.T. Lanthanide amidinates and guanidinates in catalysis and materials science: A continuing success story. Chem. Soc. Rev. 2012, 41, 7657–7672.
- Trifonov, A.A. Guanidinate and amidopyridinate rare-earth complexes: Towards highly reactive alkyl and hydrido species. Coord. Chem. Rev. 2010, 254, 1327–1347.
- Zhou, Y.; Yap, G.P.A.; Richeson, D.S. N-Substituted Guanidinate Anions as Ancillary Ligands in Organolanthanide Chemistry. Synthesis and Characterization of 2SmCH(SiMe3)2. Organometallics 1998, 17, 4387–4391.
- Lu, Z.; Yap, G.P.A.; Richeson, D.S. Tetrasubstituted Guanidinate Anions as Supporting Ligands in Organoyttrium Chemistry. Organometallics 2001, 20, 706–712.
- Lyubov, D.M.; Fukin, G.K.A.; Trifonov, A. N,N′-Diisopropyl-N߱-bis(trimethylsilyl)guanidinate Ligand as a Supporting Coordination Environment in Yttrium Chemistry. Synthesis, Structure, and Properties of Complexes YCl2(THF)2, Y(CH2SiMe3)2(THF)2, and Y2(THF). Inorg. Chem. 2007, 46, 11450–11456.
- Trifonov, A.A.; Lyubov, D.M.; Fedorova, E.A.; Fukin, G.K.; Schumann, H.; Mühle, S.; Hummert, M.; Bochkarev, M.N. Chloro, Alkyl and Aryl Complexes of Rare Earth Metals Supported by Bulky Tetrasubstituted Guanidinate Ligands. Eur. J. Inorg. Chem. 2006, 747–756.
- Trifonov, A.A.; Lyubov, D.M.; Fukin, G.K.; Baranov, E.V.; Kurskii, Y.A. Alkylyttrium Complexes Supported by N,N′-Dicyclohexyl-N′-bis(trimethylsilyl)guanidinate Ligands. Organometallics 2006, 25, 3935–3942.
- Zhang, J.; Cai, R.; Weng, L.; Zhou, X. Insertion of a Carbodiimide into the Ln−N σ-Bond of Organolanthanide Complexes. Isomerization and Rearrangement of Organolanthanides Containing Guanidinate Ligands. Organometallics 2004, 23, 3303–3308.
- Ma, L.; Zhang, J.; Cai, R.; Chen, Z.; Weng, L.; Zhou, X. Synthesis and reactivity of organolanthanide complexes containing phenothiazine ligand toward carbodiimide and isothiocyanate. J. Organomet. Chem. 2005, 690, 4926–4932.
- Pi, C.; Zhang, Z.; Pang, Z.; Zhang, J.; Luo, J.; Chen, Z.; Weng, L.; Zhou, X. Multiple N−H Bond Activation: Synthesis and Reactivity of Functionalized Primary Amido Ytterbium Complexes. Organometallics 2007, 26, 1934–1946.
- Jiang, W.; Zhang, L.; Zhang, L. Reactivity of a mixed methyl–aminobenzyl guanidinate lutetium complex towards iPrN=C=NiPr, CS2 and Ph2PH. Dalton Trans. 2022, 51, 12650–12660.
- Pi, C.; Liu, R.; Zheng, P.; Chen, Z.; Zhou, X. Selective Reaction Based on the Linked Diamido Ligands of Dinuclear Lanthanide Complexes. Inorg. Chem. 2007, 46, 5252–5259.
- Zhang, J.; Zhou, X.; Cai, R.; Weng, L. Reactivity of Organolanthanide and Organolithium Complexes Containing the Guanidinate Ligands toward Isocyanate or Carbodiimide: Synthesis and Crystal Structures. Inorg. Chem. 2005, 44, 716–722.
- Zhang, Z.; Zhang, L.; Li, Y.; Hong, L.; Chen, Z.; Zhou, X. Activation of Bis(guanidinate)lanthanide Alkyl and Aryl Complexes on Elemental Sulfur: Synthesis and Characterization of Bis(guanidinate)lanthanide Thiolates and Disulfides. Inorg. Chem. 2010, 49, 5715–5722.
- Zhang, X.; Wang, C.; Xue, M.; Zhang, Y.; Yao, Y.; Shen, Q. Synthesis and structure of samarium benzyl complex supported by bridged bis(guanidinate) ligand and its reactivity toward nitriles and phenyl isocyanate. J. Organomet. Chem. 2012, 716, 86–94.
- Zheng, P.; Hong, J.; Liu, R.; Zhang, Z.; Pang, Z.; Weng, L.; Zhou, X. Synthesis and Reactivities of Guanidinate Dianion Complexes of Heterobimetallic Lanthanide−Lithium Cp2LnLi(THF)3. Organometallics 2010, 29, 1284–1289.
- Ge, S.; Meetsma, A.; Hessen, B. Highly Efficient Hydrosilylation of Alkenes by Organoyttrium Catalysts with Sterically Demanding Amidinate and Guanidinate Ligands. Organometallics 2008, 27, 3131–3135.
- Trifonov, A.A.; Skvortsov, G.G.; Lyubov, D.M.; Skorodumova, N.A.; Fukin, G.K.; Baranov, E.V.; Glushakova, V.N. Postmetallocene Lanthanide–Hydrido Chemistry: A New Family of Complexes (Ln=Y, Nd, Sm, Gd, Yb) Supported by Guanidinate Ligands—Synthesis, Structure, and Catalytic Activity in Olefin Polymerization. Chem. Eur. J. 2006, 12, 5320–5327.
- Luo, Y.; Yao, Y.; Shen, Q. 2Ln(μ-Me)2Li(TMEDA) (Ln = Nd, Yb) as Effective Single-Component Initiators for Styrene Polymerization. Macromolecules 2002, 35, 8670–8671.
- Trifonov, A.A.; Fedorova, E.A.; Fukin, G.K.; Bochkarev, M.N. Post-Metallocene Hydridolanthanide Chemistry: 2—A Novel Lanthanide Hydride in a Non-Cyclopentadienyl Coordination Environment; Synthesis, Structure and Catalytic Activity in Olefin Polymerization. Eur. J. Inorg. Chem. 2004, 4396–4401.
- Lyubov, D.M.; Bubnov, A.M.; Fukin, G.K.; Dolgushin, F.M.; Antipin, M.Y.; Pelcé, O.; Schappacher, M.; Guillaume, S.M.; Trifonov, A.A. Hydrido Complexes of Yttrium and Lutetium Supported by Bulky Guanidinato Ligands 2 (Ln = Y, Lu): Synthesis, Structure, and Reactivity. Eur. J. Inorg. Chem. 2008, 2090–2098.
- Luo, Y.; Yao, Y.; Shen, Q.; Yu, K.; Weng, L. Synthesis and Characterization of Lanthanide(III) Bis(guanidinate) Derivatives and the Catalytic Activity of Methyllanthanide Bis(guanidinate) Complexes for the Polymerization of ϵ-Caprolactone and Methyl Methacrylate. Eur. J. Inorg. Chem. 2003, 318–323.
- Beer, S.M.J.; Krusenbaum, A.; Winter, M.; Vahlas, C.; Devi, A. Study on Structural and Thermal Characteristics of Heteroleptic Yttrium Complexes as Potential Precursors for Vapor Phase Deposition. Eur. J. Inorg. Chem. 2020, 3587–3596.
More
Information
Subjects:
Chemistry, Inorganic & Nuclear
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
18 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No