Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maša Čater | -- | 2929 | 2022-10-17 10:41:25 | | | |
| 2 | Vivi Li | -1 word(s) | 2928 | 2022-10-18 03:32:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Čater, M.; Hölter, S.M. A Pathophysiological Intersection of Diabetes and Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/29649 (accessed on 12 June 2026).
Čater M, Hölter SM. A Pathophysiological Intersection of Diabetes and Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/29649. Accessed June 12, 2026.
Čater, Maša, Sabine M. Hölter. "A Pathophysiological Intersection of Diabetes and Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/29649 (accessed June 12, 2026).
Čater, M., & Hölter, S.M. (2022, October 17). A Pathophysiological Intersection of Diabetes and Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/29649
Čater, Maša and Sabine M. Hölter. "A Pathophysiological Intersection of Diabetes and Alzheimer’s Disease." Encyclopedia. Web. 17 October, 2022.
Copy Citation
Diabetes is among the most prevalent diseases of the modern world and is strongly linked to an increased risk of numerous neurodegenerative disorders, although the exact pathophysiological mechanisms are not clear yet. Insulin resistance is a serious pathological condition, connecting type 2 diabetes, metabolic syndrome, and obesity. Insulin resistance has been proven to be connected also to cognitive decline and dementias, including the most prevalent form, Alzheimer’s disease. The relationship between diabetes and Alzheimer’s disease regarding pathophysiology is so significant that it has been proposed that some presentations of the condition could be termed type 3 diabetes.
diabetes
insulin sensitivity
Alzheimer’s disease
brain
cognition
depression
1. Introduction
The incidence of age-related metabolic and neurodegenerative diseases is increasing as the world population is aging [1][2]. Many studies confirmed that patients with impaired glucose tolerance and diabetes have an increased risk of developing Alzheimer’s disease (AD) compared to healthy individuals [3][4][5]. The association between diabetes and its effects on the development of retinopathy, neuropathy, nephropathy, and cardiovascular diseases is well established [6]. However, the underlying processes of how diabetes impacts the brain, cognition, and mental health are not yet fully understood. Therefore, in this entry, researchers gathered up-to-date literature about the effects of impaired glucose metabolism on the brain, cognition, and mental health, and about the intersection of symptoms of diabetes and AD.
AD, the most common cause of dementia, is a progressive brain disorder that causes memory deficits and destroys cognitive functioning. A genetic mutation is proposed to be the cause for early-onset AD, while a sum of genetic, environmental and lifestyle factors are considered to trigger the late-onset AD [7]. Changes in the brain, that occur in the early stages of the disorder, include an abnormal buildup of proteins that form amyloid plaques and tau tangles. This results in neuron malfunctioning and a loss of connections between neurons, and eventually, in neuron death [8]. The damage in the brain is initiated in the hippocampus and the entorhinal cortex, both essential in memory formation. Therefore, the first signs of AD include memory problems and mild cognitive impairment. When damage spreads to other parts of the brain, it causes shrinkage of the brain. Thus, in the mild stage of AD, greater memory loss occurs with additional personality and behavior changes. Moreover, in moderate AD, language, reasoning, and sensory processing are affected by the damage in the brain. Hallucinations, delusions, and paranoia can occur at this stage as well. When AD progresses to a severe stage, with plaques and tangles that are wide-spread throughout the brain, communication and independent self-care become greatly affected [9].
AD and diabetes share many different pathologies, such as hyperglycemia, glucose intolerance, hyperinsulinemia, insulin resistance, adiposity, hypertension, atherosclerosis, and cognitive impairment [10][11][12]. The main bases for their development are proposed to be the malfunctioning of insulin signaling [13] with glucose metabolism disorders, deficits in mitochondrial activity, and cholesterol-associated pathologies—these are some of the many additional causes that have been observed by diabetes researchers and neuroscientists in recent decades [6][14][15][16][17]. In the following sections, researchers will look at each of the proposed factors causing diabetes and/or AD and find their correlations. Moreover, researchers will discuss the most frequently used animal models in diabetes and AD studies, which enabled the understanding of the mechanism of action of compromised insulin activity-related pathologies. This entry is based on a PubMed search using combinations of the terms, «diabetes», «Alzheimer’s disease», «animal model», «mouse», «rat», «insulin» and «brain». Up-to-date references were selected by the date of publication (last 10–15 years, with exceptions) and by their main discovery conclusions so they fitted to the scope of this entry in at least one of the aspects (diabetes or AD).
2. The Importance of Brain Insulin
The brain is the most energy-demanding organ that relies on glucose for fuel. Most of its energy expenditure is needed for maintaining the potential difference across the membranes of nerve cells—for dendritic and axonal transport, and tissue repair. The brain uses several pathways for gathering glucose. Glucose enters the brain by insulin-insensitive facilitated diffusion across the blood–brain barrier (BBB), followed by entering the brain cells using insulin-independent glucose transporters [18][19] as well as insulin-regulated glucose transporters [20].
Thus, insulin, a highly active hormone and a crucial polypeptide in the body, plays an important role in the brain (Figure 1). Like glucose, insulin also crosses the BBB, and when in the brain, insulin binds to insulin receptors on neurons and glial cells; there, its main function is to modulate glucose transfer into different brain cells for maintaining the normal functioning of the brain [21]. Additionally, insulin in the brain contributes to the control of nutrient homeostasis, reproduction, cognition, and memory, as well as to neurotrophic, neuromodulatory [22], and neuroprotective effects.
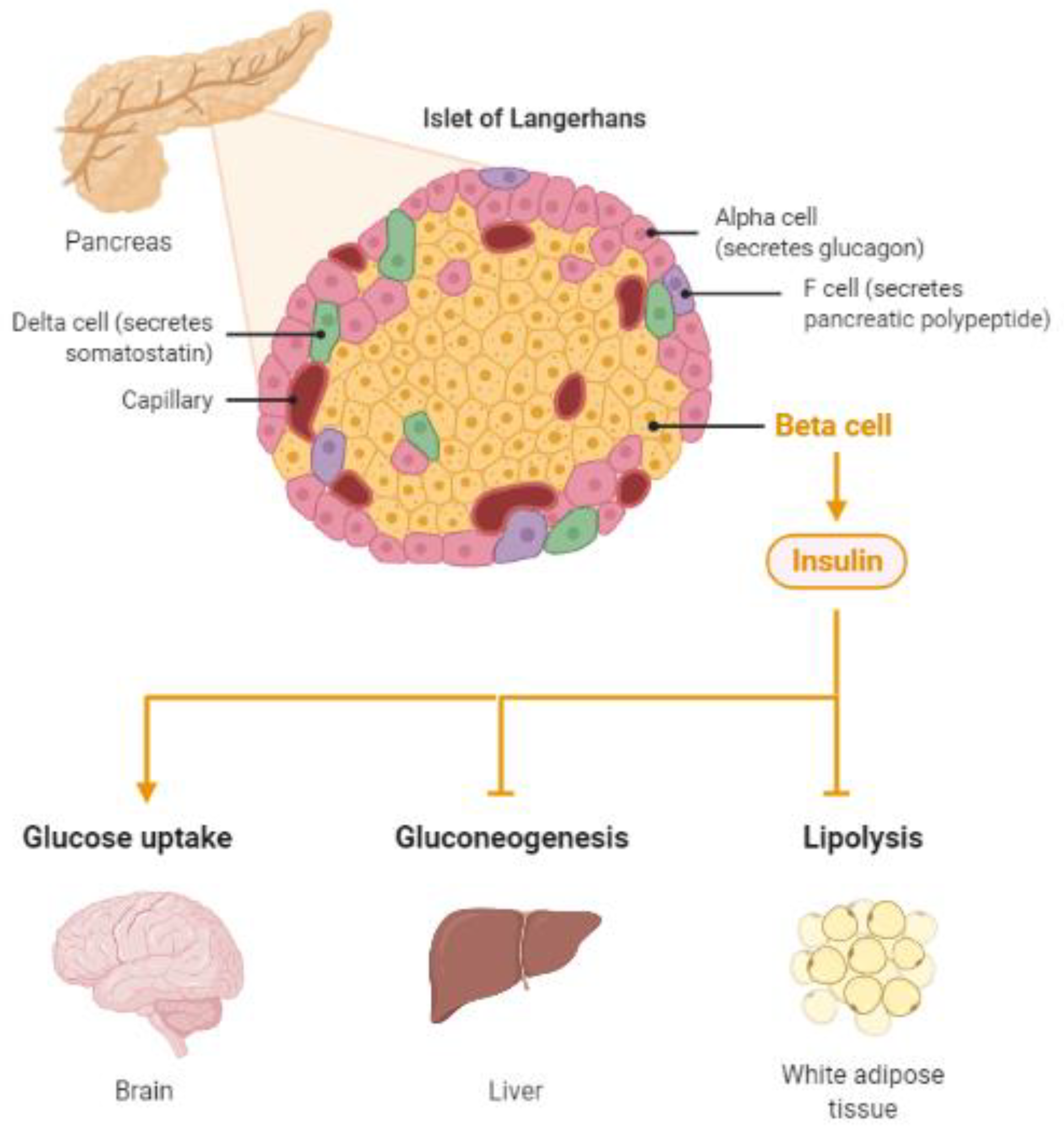
Figure 1. Insulin, a protein secreted by the pancreas, plays a major role in energy homeostasis of the body. Insulin pathologies are directly involved in the development of diabetes and Alzheimer’s disease (Created by BioRender.com, accessed on 23 August 2021).
A third option for brain cells to gather glucose has been discovered in recent decades [23]. A fascinating detection of insulin mRNA transcripts in the brain revealed that the brain is capable of synthesizing insulin on its own [22][24][25].
3. Involvement of Diabetes in the Development of Alzheimer’s Disease
3.1. Abnormal Protein Processing
The main feature of AD is abnormal protein processing in the brain, through which amyloid-β plaques and neurofibrillary tangles are formed, causing morphological pathologies in the brain tissue due to disintegrated microtubules, synaptic impairment, and neuronal apoptosis, thus, resulting in cognitive impairments and several psychopathologies (Figure 2).
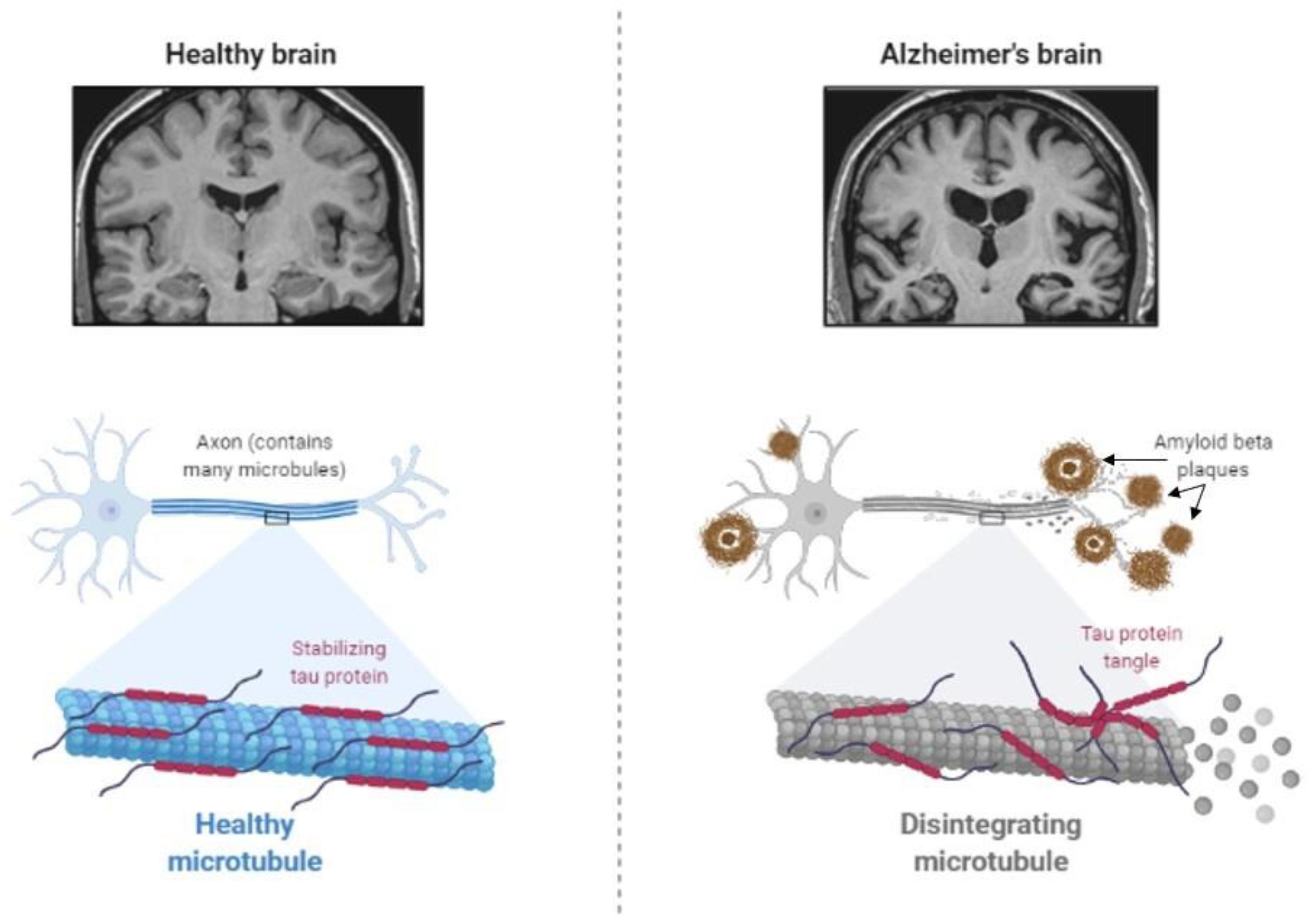
Figure 2. Abnormal protein processing occurs in the brain of patients with Alzheimer’s disease, which leads to the disintegration of microtubules and formation of tau aggregates and amyloid-β plaques, causing morphological pathologies, several cognitive impairments, and behavioral changes (Created by BioRender.com, accessed on 23 August 2021).
Diabetes has been shown to be involved in the development of AD by affecting abnormal protein processing. Disturbed insulin signaling, associated with type 2 diabetes, affects the expression and metabolism of amyloid-β [26]. Moreover, abnormally phosphorylated tau protein, which induces the instability of neuronal microtubules and apoptosis of neurons in AD [27][28], has been observed in animal models of diabetes. Increased tau phosphorylation has been observed in type 1 and type 2 diabetes of mouse and rat models [29][30][31][32][33], as well as in humans with type 2 diabetes [34].
3.2. Deficient Insulin Signaling
Dysfunctional insulin receptor signaling is known to affect the expression and metabolism of amyloid-β and tau protein [26] and their clearance [35]. Insulin receptors are proposed to regulate synaptic activity as well; therefore, the malfunctioning of the receptors could cause neurodegeneration [36]. Moreover, insulin resistance in connection with hyperinsulinemia induces the accumulation of amyloid-β due to the lack of available insulin-degrading enzymes (IDEs). Normally, insulin and amyloid-β are both degraded by IDE. When insulin levels are increased, such as in type 2 diabetes, insulin uses the majority of IDE, and undegraded amyloid-β starts to accumulate in neurons [37]. As a possible treatment, insulin sensitizers have been tested in rodents [38] and early AD patients [39], with positive results in improving cognitive performances. Further studies are still needed to confirm the exact potential of insulin sensitizers for AD treatment. Apart from increased amyloid-β accumulation in neurons, hyperinsulinemia causes tau hyperphosphorylation in primary cortical neurons and hippocampal neurons [40][41][42], provoking their degeneration.
Alterations in insulin receptor signaling in type 2 diabetes and AD develop due to changes in both major signaling pathways. The mitogen-activated protein kinase (MAPK) pathway, required for cell proliferation, differentiation, and apoptosis [43], is accelerated in the brain of patients with AD [44]. The expression of MAPK co-localizes with aggregated tau in the hippocampus and cortical regions in AD brains, indicating that MAPK signaling is also involved in tau phosphorylation, synaptic plasticity, and neuroinflammation [45][46]. With respect to diabetes, Dusp8, which codes for a dual-specificity phosphatase involved in MAPK signaling and is predominantly expressed in the brain, was implicated by genome-wide association studies as a type 2 diabetes risk gene. There is evidence that Dusp8 can have sex-specific effects in mice and men on hypothalamic insulin resistance, hippocampal size, and cognitive, emotional, and hedonic behaviors [47][48][49]. The second insulin receptor signaling pathway, the Akt pathway—which is responsible for cell growth and survival, protein synthesis, and inhibition of the glycogen synthase kinase-3β (GSK-3β) enzyme [50][51][52][53][54]—is also affected by both AD and type 2 diabetes [55]. GSK-3β in the hippocampus and cortex is important for glycogenesis and glucose clearance. In normal conditions, its activity is inhibited by phosphorylation by insulin signaling via an insulin receptor. However, in type 2 diabetes, elevated activity of GSK-3β is proposed to trigger the reduction in glucose clearance by developing insulin resistance [54]. Moreover, increased GSK-3β activity is thought to result in increased amyloid-β production and tau phosphorylation [53][56]. Experiments in AD animal models and cell cultures have shown that GSK-3β is a good target for treatment development, as inhibiting GSK-3β successfully slowed down neurodegeneration [56][57].
3.3. The Cholinergic Hypothesis
Acetylcholine is a neurotransmitter that is involved in cholinergic neurotransmission. It is used by cholinergic neurons and has an important role in the peripheral and central nervous system, as cholinergic neurons are critical for cognition and memory. AD patients present with a continuous decline of cholinergic neurotransmission in their brain [58]. This occurs due to a decrease in the production of acetylcholine as well as the hydrolysis of acetylcholine. A mechanism of action has been proposed with a cholinergic hypothesis, which suggests that insulin plays an important role in acetylcholine production [59]. In case of hypoinsulinemia, less acetylcholine is produced due to a reduced expression of choline acetyltransferase (Figure 3). This direct effect of insulin on acetylcholine production has additionally strengthened the link between AD development, insulin malfunction, and diabetes; thus, AD has recently been considered a neuroendocrine disease and has been referred to as type 3 diabetes, possessing characteristics of type I and type II diabetes [59][60][61][62].
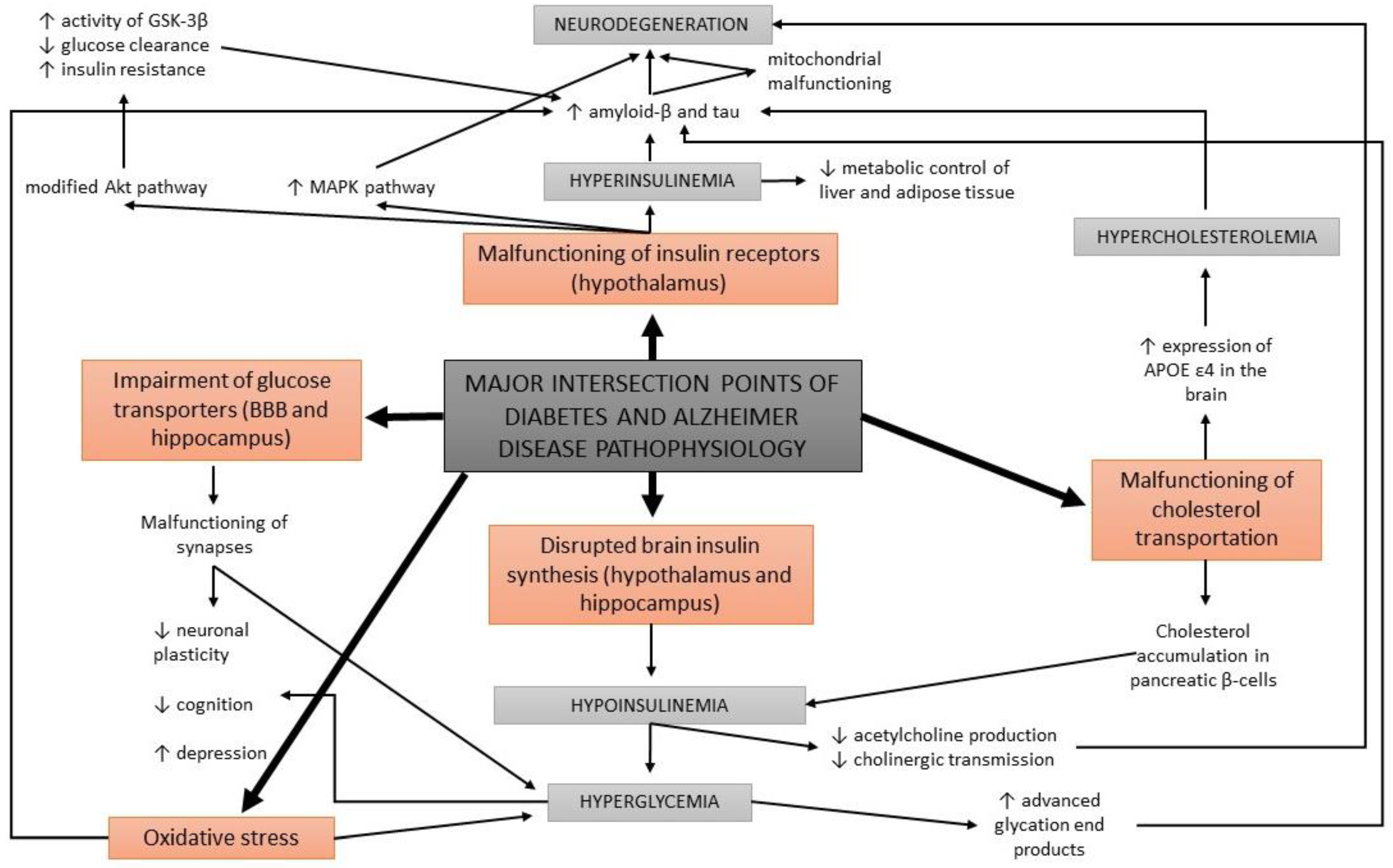
Figure 3. A schematic overview of the major intersection points of diabetes and Alzheimer’s disease pathophysiology.
3.4. Glucose Metabolism Disorders
Apart from the impairments in brain insulin production and signaling, additional conditions are found at the intersection of diabetes and AD, such as oxidative stress and the formation of advanced glycation end products. Abnormal glucose metabolism and oxidative stress trigger the formation of advanced glycation end products which cause brain damage [14]. Advanced glycation end products are formed by the glycation of proteins or lipids in case of chronic hyperglycemia, and present a useful biomarker for degenerative diseases such as diabetes and AD. In normal aging, the formation of these molecules occurs in low levels, while it is greatly accelerated in patients with diabetes and AD [63][64]. Moreover, advanced glycation end products were found to induce the glycation of amyloid-β and tau, resulting in their formation and aggregation [65] (Figure 3). An increased expression of receptors for advanced glycation end products in neurons was determined in diabetic mice with impaired cognition [66], as well as at a clinical level in patients with AD and diabetes compared to non-diabetic AD patients [67].
3.5. Oxidative Stress
Impaired glucose metabolism has other effects in the body as well. It causes an accelerated production of free radicals, which results in oxidative stress in cells. Oxidative stress is well known to contribute to the development of diabetes and its neuropathies [68][69]. It has been indicated that oxidative cell damage occurs early in the development of AD [15], as increased concentrations of oxidized proteins in the hippocampus and in the frontal and parietal lobes were determined in patients with only a mild cognitive impairment. Oxidative stress is strongly linked to amyloid-β accumulation. Preclinical research in mice showed that antioxidant capacity decreases first, followed by an increase in lipid peroxidation, and finally, results in AD development [70][71].
Moreover, oxidative stress triggers local inflammations in the brain [72]. Some studies report that the use of anti-inflammatory drugs can decrease the risk of AD development, while others report no beneficial effects of taking these agents for treating AD [73][74][75].
3.6. Deficits in Mitochondrial Activity
Another factor correlating diabetes to AD is mitochondrial malfunctioning [6][16][55]. Mitochondrial activity is essential for normal neuronal functioning regarding ATP synthesis and for controlling calcium homeostasis [76]. While the efficiency of calcium homeostasis regulation decreases in the brain with normal aging, an increased calcium uptake by mitochondria has been observed among AD pathologies [77][78], as well as a decrease in mitochondrial mass and an increase in mitochondrial DNA in the cytoplasm [79]. Excessive calcium uptake triggers an increase in the level of reactive oxygen species and inhibition of ATP synthesis, resulting in neuronal degeneration and apoptosis. The exact mechanism of action regarding how mitochondrial malfunctioning is correlated to AD is not yet fully understood. However, it is proposed that mitochondrial electron transport is negatively affected by amyloid-β [80], which further on leads to mitochondrial malfunction and dysregulation of calcium homeostasis. Increased levels of intracellular calcium have been found to co-localize with neurofibrillary tangles and amyloid-β aggregates [78][81][82].
Interestingly, as excessive calcium uptake by mitochondria causes damage in neurons in AD, it initiates similar damage also in pancreatic cells, causing insulin malfunctions that lead to diabetes pathologies [83]. High levels of calcium in pancreatic β-cells are thought to trigger the malfunctioning of insulin secretion [84]. Insulin deficiency and oxidative stress due to a lower antioxidant capacity of neuronal mitochondria have been observed in type 1 diabetic rats [85], and these defects in mitochondrial DNA are determined to be inheritable. Moreover, in type 2 diabetes, a similar low antioxidant activity of mitochondria has been observed. However, in this type of diabetes, obesity is usually present, which is also known to be associated with smaller mitochondria and reduced energetic capacity [86].
3.7. Cholesterol-Associated Pathologies
The malfunctioning of cholesterol transportation within the circulatory system has been observed in diabetes and AD, yet the details of the underlying processes are unclear. In the case of diabetes, cholesterol has been found to be accumulated within pancreatic β-cells, causing a decrease in insulin secretion [87]. In AD mouse models, cholesterol has been determined at the same locations as amyloid-β plaques and tau proteins [88]. Therefore, it has been proposed that cholesterol is directly involved in the formation of protein abnormalities in AD (Figure 3).
Normal blood lipid levels are maintained by apolipoprotein E, which is expressed mainly in the brain and liver. Some alleles (APOE ε4) of the gene for apolipoprotein E result in the development of hypercholesterolemia and have been found in 40% of AD patients [55]. Additionally, the risk for AD development associated with APOE ε4 is doubled by diabetes [89]. Apolipoprotein E, synthesized from APOE ε4, is linked to abnormal protein processing, which is present in AD patients; in addition, apolipoprotein E is able to cooperate with amyloid-β aggregates and it promotes the phosphorylation of tau in neurons, inducing neurodegeneration [17][90].
4. Insulin Effects on Cognition and Mental Health
4.1. Cognitive Changes
Cognitive changes in people with dementia are well documented and studied. Recently, diabetes has received significant attention in psychiatry due to its high mutual frequency with mood disorders and cognitive impairment. Several symptoms of AD, such as cognitive dysfunctions regarding attention, memory, vocabulary, information processing, motor strength and speed, visual-motor and spatial skills as well as impaired general intelligence, were observed in patients with type 1 and type 2 diabetes [55][91]. Deficits in spatial learning and long-term potentiation in the hippocampus, which are important for memory formation, have been observed in patients with type 1 diabetes. Moreover, the most prevalent form of diabetes, type 2 diabetes, has been determined to trigger early cognitive and mental changes in a similar way as type 1 diabetes. While in type 1 diabetes the major problem causing physiological and cognitive deficits is insulin deficiency, in type 2 diabetes it is the malfunctioning of insulin receptor activity that has enormous consequences on the brain. Hence, insulin resistance, hyperinsulinemia, and impaired insulin signaling have been determined to cause many cognitive pathologies (Figure 4) [50][55].
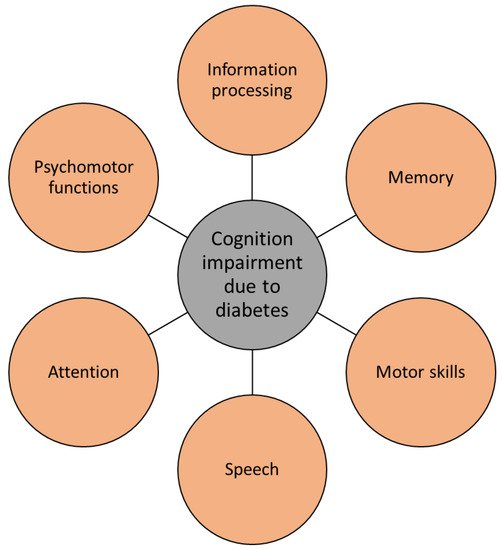
Figure 4. Diabetes causes several cognitive impairments, typical for Alzheimer’s disease.
The age of onset of diabetes, its duration, and degree of glycemic control are the main factors affecting how severe cognitive dysfunction will occur [92][93]. The degree of cognitive impairment progresses with long-lasting diabetes and poorly maintained glycemic control, and with the presence of diabetic complications such as depression and hypertension [21].
4.2. Mental Changes
Alzheimer’s disease is accompanied by depression and anxiety, which are the most prevalent mood disorders [94]. Diabetes often co-occurs with depression and anxiety, suggesting that insulin malfunctioning can play a role in mental changes. In association with the various effects on different brain cells, insulin has been shown to affect not only cognition, but also mood and psychiatric functioning [21]. The importance of brain insulin resistance is clearly seen in neuronal insulin receptor knock-out mice, which display spontaneous and life-long depressive-like and anxiety-like phenotypes [95].
The idea of influencing emotional behaviors with insulin dates back a century, when psychiatrists used insulin to cure mental illnesses by inducing a coma or as a shock treatment [96]. Now, researchers know that insulin receptors are highly present in the limbic region of the brain, where reward-based functioning, motivation, and emotions derive from. Many studies have proposed that diabetes and the development of mood disorders and their increased severity are causally linked [97]. One of the possibilities for the mechanism of action is the insulin modulation of brain serotonergic neurons and their neurotransmission, resulting in the development of anxiety and depressive symptoms [98]. Therefore, it is largely accepted that the risk of developing mood disorders is significantly increased in diabetic patients. Interestingly, there is a bidirectional correlation, as a 60% increase in risk for developing type 2 diabetes has been observed in depressed patients [99], yet a detailed mechanism of this correlation is not fully understood. Anhedonia is one of the domains of depression, defined as an incapacity to feel pleasure. It is suggested that anhedonia is associated with poor glycemic control in patients with type 2 diabetes. However, in patients with type 1 diabetes, a negative correlation between anhedonia symptoms and glycemic control was reported [100][101]. Further research is needed to clarify the mechanism of action which connects anhedonia with diabetes and AD.
References
- Kelly, T.; Yang, W.; Chen, C.-S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437.
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321.
- Brands, A.M.; Biessels, G.J.; De Haan, E.H.; Kappelle, L.J.; Kessels, R.P. The effects of type 1 diabetes on cognitive performance: A meta-analysis. Diabetes Care 2005, 28, 726–735.
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74.
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481.
- Čater, M.; Križančić Bombek, L. Protective Role of Mitochondrial Uncoupling Proteins against Age-Related Oxidative Stress in Type 2 Diabetes Mellitus. Antioxidants 2022, 11, 1473.
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annu. Rev. Pathol. 2022, 17, 73–99.
- Liu, R.-M. Aging, cellular senescence, and Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 1989.
- Cammisuli, D.M.; Cipriani, G.; Castelnuovo, G. Technological solutions for diagnosis, management and treatment of Alzheimer0s disease-related symptoms: A structured review of the recent scientific literature. Int. J. Environ. Res. Public Health 2022, 19, 3122.
- Ekblad, L.L.; Rinne, J.O.; Puukka, P.; Laine, H.; Ahtiluoto, S.; Sulkava, R.; Viitanen, M.; Jula, A. Insulin resistance predicts cognitive decline: An 11-year follow-up of a nationally representative adult population sample. Diabetes Care 2017, 40, 751–758.
- Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071.
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338.
- Haan, M.N. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166.
- Biessels, G.J.; van der Heide, L.P.; Kamal, A.; Bleys, R.L.; Gispen, W.H. Ageing and diabetes: Implications for brain function. Eur. J. Pharmacol. 2002, 441, 1–14.
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248.
- Moreira, P.I.; Santos, M.S.; Seiça, R.; Oliveira, C.R. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J. Neurol. Sci. 2007, 257, 206–214.
- Belinson, H.; Lev, D.; Masliah, E.; Michaelson, D.M. Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J. Neurosci. 2008, 28, 4690–4701.
- Benarroch, E.E. Brain glucose transporters: Implications for neurologic disease. Neurology 2014, 82, 1374–1379.
- Rebelos, E.; Rinne, J.O.; Nuutila, P.; Ekblad, L.L. Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline—Does Insulin Have Anything to Do with It? A Narrative Review. J. Clin. Med. 2021, 10, 1532.
- Reno, C.M.; Puente, E.C.; Sheng, Z.; Daphna-Iken, D.; Bree, A.J.; Routh, V.H.; Kahn, B.B.; Fisher, S.J. Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemic counterregulation. Diabetes 2017, 66, 587–597.
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181.
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161.
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80.
- Kuwabara, T.; Kagalwala, M.N.; Onuma, Y.; Ito, Y.; Warashina, M.; Terashima, K.; Sanosaka, T.; Nakashima, K.; Gage, F.H.; Asashima, M. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol. Med. 2011, 3, 742–754.
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012, 16, 723–737.
- Li, L.; Hölscher, C. Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res. Rev. 2007, 56, 384–402.
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729.
- Chung, C.-W.; Song, Y.-H.; Kim, I.-K.; Yoon, W.-J.; Ryu, B.-R.; Jo, D.-G.; Woo, H.-N.; Kwon, Y.-K.; Kim, H.-H.; Gwag, B.-J. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol. Dis. 2001, 8, 162–172.
- Kim, B.; Backus, C.; Oh, S.; Hayes, J.M.; Feldman, E.L. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 2009, 150, 5294–5301.
- Li, Z.-G.; Zhang, W.; Sima, A.A. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007, 56, 1817–1824.
- Clodfelder-Miller, B.J.; Zmijewska, A.A.; Johnson, G.V.; Jope, R.S. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 2006, 55, 3320–3325.
- Planel, E.; Tatebayashi, Y.; Miyasaka, T.; Liu, L.; Wang, L.; Herman, M.; Yu, W.H.; Luchsinger, J.A.; Wadzinski, B.; Duff, K.E. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 2007, 27, 13635–13648.
- Jolivalt, C.; Lee, C.; Beiswenger, K.; Smith, J.; Orlov, M.; Torrance, M.; Masliah, E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: Parallels with Alzheimer’s disease and correction by insulin. J. Neurosci. Res. 2008, 86, 3265–3274.
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J. Neurochem. 2009, 111, 242–249.
- Zhao, W.-Q.; Lacor, P.N.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric Aβ. J. Biol. Chem. 2009, 284, 18742–18753.
- Zhao, W.-Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 2001, 177, 125–134.
- Gasparini, L.; Xu, H. Potential roles of insulin and IGF-1 in Alzheimer’s disease. Trends Neurosci. 2003, 26, 404–406.
- Pedersen, W.A.; McMillan, P.J.; Kulstad, J.J.; Leverenz, J.B.; Craft, S.; Haynatzki, G.R. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 2006, 199, 265–273.
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958.
- Lesort, M.; Johnson, G. Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience 2000, 99, 305–316.
- Freude, S.; Plum, L.; Schnitker, J.; Leeser, U.; Udelhoven, M.; Krone, W.; Bruning, J.C.; Schubert, M. Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes 2005, 54, 3343–3348.
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 2003, 23, 7084–7092.
- Pearson, L.L.; Castle, B.E.; Kehry, M.R. CD40-mediated signaling in monocytic cells: Up-regulation of tumor necrosis factor receptor-associated factor mRNAs and activation of mitogen-activated protein kinase signaling pathways. Int. Immunol. 2001, 13, 273–283.
- Hensley, K.; Floyd, R.A.; Zheng, N.Y.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R. p38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 1999, 72, 2053–2058.
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568.
- Kelleher, I.; Garwood, C.; Hanger, D.P.; Anderton, B.H.; Noble, W. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 2007, 103, 2256–2267.
- Baumann, P.; Schriever, S.; Kullmann, S.; Zimprich, A.; Feuchtinger, A.; Amarie, O.; Peter, A.; Walch, A.; Gailus-Durner, V.; Fuchs, H.; et al. Dusp8 affects hippocampal size and behavior in mice and humans. Sci. Rep. 2019, 9, 19483.
- Baumann, P.; Schriever, S.; Kullmann, S.; Zimprich, A.; Peter, A.; Gailus-Durner, V.; Fuchs, H.; Hrabe de Angelis, M.; Wurst, W.; Tschöp, M.; et al. Diabetes type 2 risk gene Dusp8 is associated with altered sucrose reward behavior in mice and humans. Brain Behav. 2021, 11, e01928.
- Schriever, S.; Kabra, D.; Pfuhlmann, K.; Baumann, P.; Baumgart, E.; Nagler, J.; Seebacher, F.; Harrison, L.; Irmler, M.; Kullmann, S.; et al. Type 2 diabetes risk gene Dusp8 regulates hypothalamic Jnk signaling and insulin sensitivity. J. Clin. Investig. 2020, 130, 6093–6108.
- de la Monte, S.M.; Wands, J.R. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer’s disease. J. Alzheimer’s Dis. 2005, 7, 45–61.
- Brazil, D.P.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664.
- Tremblay, M.L.; Giguère, V. Phosphatases at the heart of FoxO metabolic control. Cell Metab. 2008, 7, 101–103.
- Balaraman, Y.; Limaye, A.; Levey, A.; Srinivasan, S. Glycogen synthase kinase 3β and Alzheimer’s disease: Pathophysiological and therapeutic significance. Cell. Mol. Life Sci. CMLS 2006, 63, 1226–1235.
- Lee, J.; Kim, M.-S. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract. 2007, 77, S49–S57.
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559.
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 2003, 423, 435–439.
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115.
- Kroner, Z. The Relationship between Alzheimer’s Disease and Diabetes: Type 3 Diabetes. Altern. Med. Rev. 2009, 14, 373–379.
- Abeysekera, W.K.S.M.; Ratnasooriya, W.D.; Abeysekera, W.P.K.M.; Premakumara, G.A.S. Anti-Acetylcholinesterase Activity of Commercially Important Ceylon Black Tea (Camellia sinensis L.) Grades Belonging to Different Elevations: Potential Natural Product for Type 3 Diabetes Management? Asian Food Sci. J. 2021, 20, 36–46.
- Leszek, J.; Trypka, E.; Tarasov, V.V.; Ashraf, G.M.; Aliev, G. Type 3 diabetes mellitus: A novel implication of Alzheimers disease. Curr. Top. Med. Chem. 2017, 17, 1331–1335.
- de la Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes—Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113.
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146.
- Brownlee, M.M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234.
- Ledesma, M.D.; Bonay, P.; Colaço, C.; Avila, J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J. Biol. Chem. 1994, 269, 21614–21619.
- Toth, C.; Schmidt, A.M.; Tuor, U.I.; Francis, G.; Foniok, T.; Brussee, V.; Kaur, J.; Yan, S.F.; Martinez, J.A.; Barber, P.A. Diabetes, leukoencephalopathy and rage. Neurobiol. Dis. 2006, 23, 445–461.
- Gironès, X.; Guimerà, A.; Cruz-Sánchez, C.-Z.; Ortega, A.; Sasaki, N.; Makita, Z.; Lafuente, J.V.; Kalaria, R.; Cruz-Sánchez, F.F. Nϵ-Carboxymethyllysine in brain aging, diabetes mellitus, and Alzheimer’s disease. Free Radic. Biol. Med. 2004, 36, 1241–1247.
- Russell, J.W.; Berent-Spillson, A.; Vincent, A.M.; Freimann, C.L.; Sullivan, K.A.; Feldman, E.L. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 2008, 30, 420–429.
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628.
- Matsuoka, Y.; Picciano, M.; La Francois, J.; Duff, K. Fibrillar β-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer’s disease. Neuroscience 2001, 104, 609–613.
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057.
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421.
- Szekely, C.A.; Thorne, J.E.; Zandi, P.P.; Ek, M.; Messias, E.; Breitner, J.C.; Goodman, S.N. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 2004, 23, 159–169.
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J.; et al. Effects of Rofecoxib or Naproxen vs. Placebo on Alzheimer Disease Progression. JAMA 2003, 289, 2819.
- Reines, S.; Block, G.; Morris, J.; Liu, G.; Nessly, M.; Lines, C.; Norman, B.; Baranak, C. Rofecoxib: No effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 2004, 62, 66–71.
- Rizzuto, R.; Pinton, P.; Brini, M.; Chiesa, A.; Filippin, L.; Pozzan, T. Mitochondria as biosensors of calcium microdomains. Cell Calcium 1999, 26, 193–200.
- Blass, J.; Gibson, G. The role of oxidative abnormalities in the pathophysiology of Alzheimer’s disease. Rev. Neurol. 1991, 147, 513–525.
- Nixon, R.A.; Saito, K.I.; Grynspan, F.; Griffin, W.R.; Katayama, S.; Honda, T.; Mohan, P.S.; Shea, T.B.; Beermann, M. Calcium-Activated Neutral Proteinase (Calpain) System in Aging and Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1994, 747, 77–91.
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023.
- Crouch, P.J.; Blake, R.; Duce, J.A.; Ciccotosto, G.D.; Li, Q.-X.; Barnham, K.J.; Curtain, C.C.; Cherny, R.A.; Cappai, R.; Dyrks, T. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-β1-42. J. Neurosci. 2005, 25, 672–679.
- Johnson, G.V.; Cox, T.M.; Lockhart, J.P.; Zinnerman, M.D.; Miller, M.L.; Powers, R.E. Transglutaminase activity is increased in Alzheimer’s disease brain. Brain Res. 1997, 751, 323–329.
- Querfurth, H.W.; Selkoe, D.J. Calcium ionophore increases amyloid. beta. Peptide production by cultured cells. Biochemistry 1994, 33, 4550–4561.
- Levy, J.; Gavin, J.R., III; Sowers, J.R. Diabetes mellitus: A disease of abnormal cellular calcium metabolism? Am. J. Med. 1994, 96, 260–273.
- Levy, J.; Zemel, M.B.; Sowers, J.R. Role of cellular calcium metabolism in abnormal glucose metabolism and diabetic hypertension. Am. J. Med. 1989, 87, S7–S16.
- Moreira, P.I.; Santos, M.S.; Sena, C.; Seiça, R.; Oliveira, C.R. Insulin protects against amyloid β-peptide toxicity in brain mitochondria of diabetic rats. Neurobiol. Dis. 2005, 18, 628–637.
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950.
- Ishikawa, M.; Iwasaki, Y.; Yatoh, S.; Kato, T.; Kumadaki, S.; Inoue, N.; Yamamoto, T.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N. Cholesterol accumulation and diabetes in pancreatic β-cell-specific SREBP-2 transgenic mice: A new model for lipotoxicity. J. Lipid Res. 2008, 49, 2524–2534.
- Burns, M.P.; Noble, W.J.; Olm, V.; Gaynor, K.; Casey, E.; LaFrancois, J.; Wang, L.; Duff, K. Co-localization of cholesterol, apolipoprotein E and fibrillar Aβ in amyloid plaques. Mol. Brain Res. 2003, 110, 119–125.
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262.
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.-Q.; Xu, Q.; Fish, J.D.; Wyss-Coray, T.; Buttini, M.; Mucke, L. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004, 24, 2527–2534.
- Miles, W.; Root, H. Psychologic tests applied to diabetic patients. Arch. Intern. Med. 1922, 30, 767–777.
- Awad, N.; Gagnon, M.; Messier, C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. J. Clin. Exp. Neuropsychol. 2004, 26, 1044–1080.
- Strachan, M.W.; Deary, I.J.; Ewing, F.M.; Frier, B.M. Is type II diabetes associated with an increased risk of cognitive dysfunction?: A critical review of published studies. Diabetes Care 1997, 20, 438–445.
- Ferretti, L.; McCurry, S.M.; Logsdon, R.; Gibbons, L.; Teri, L. Anxiety and Alzheimer’s Disease. J. Geriatr. Psychiatry Neurol. 2001, 14, 52–58.
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468.
- James, F.E. Insulin treatment in psychiatry. Hist. Psychiatry 1992, 3, 221–235.
- Nouwen, A.; Winkley, K.; Twisk, J.; Lloyd, C.E.; Peyrot, M.; Ismail, K.; Pouwer, F. Type 2 diabetes mellitus as a risk factor for the onset of depression: A systematic review and meta-analysis. Diabetologia 2010, 53, 2480–2486.
- Martin, H.; Bullich, S.; Guiard, B.P.; Fioramonti, X. The impact of insulin on the serotonergic system and consequences on diabetes-associated mood disorders. J. Neuroendocrinol. 2021, 33, e12928.
- Mezuk, B.; Eaton, W.W.; Albrecht, S.; Golden, S.H. Depression and type 2 diabetes over the lifespan: A meta-analysis. Diabetes Care 2008, 31, 2383–2390.
- Ehrmann, D.; Schmitt, A.; Reimer, A.; Haak, T.; Kulzer, B.; Hermanns, N. The affective and somatic side of depression: Subtypes of depressive symptoms show diametrically opposed associations with glycemic control in people with type 1 diabetes. Acta Diabetol. 2017, 54, 749–756.
- Carter, J.; Swardfager, W. Mood and metabolism: Anhedonia as a clinical target in Type 2 diabetes. Psychoneuroendocrinology 2016, 69, 123–132.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
602
Revisions:
2 times
(View History)
Update Date:
18 Oct 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No