 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Peter Kaplan | + 1467 word(s) | 1467 | 2020-10-19 09:35:22 | | | |
| 2 | Catherine Yang | -491 word(s) | 976 | 2020-11-11 02:13:18 | | |
Video Upload Options
Elevated concentration of homocysteine (Hcy) in the blood plasma, hyperhomocysteinemia (HHcy), has been implicated in various disorders, including cardiovascular and neurodegenerative diseases. Accumulating evidence indicates that pathophysiology of these diseases is linked with mitochondrial dysfunction. Mitochondria are essential for maintaining cellular homeostasis and function. The interaction between Hcy and mitochondria is complex and reactive oxygen species (ROS) seem to be important mediators of Hcy effects. Although oxidative damage to mitochondria is frequently demonstrated under HHcy, Hcy may have also beneficial effects on mitochondrial function and cell viability.
1. Introduction
Homocysteine (Hcy) is a sulfur-containing amino acid (Figure 1) formed during metabolism of methionine, an essential amino acid derived from dietary proteins. The metabolic pathway involved in Hcy formation is important due to formation of S-adenosylmethionine (SAM), a source of methyl group for methylation reactions, such as DNA methylation or formation of catecholamines. S-adenosylhomocysteine (SAH), which is formed from SAM after transfer of methyl group to various substrates, is hydrolyzed by SAH hydrolase to Hcy and adenosine. Released Hcy can be remethylated to methionine via folate and vitamin B12-dependent reaction and/or catabolized to amino acid cysteine by the vitamin B6-dependent pathway. Deficiencies in vitamins and/or enzymes involved in Hcy metabolism as well as some pathological conditions result in elevated blood plasma concentrations of Hcy, called hyperhomocysteinemia (HHcy). Hcy can induce cellular and molecular oxidative injury through reactive oxygen species (ROS) [1]. Hcy-induced oxidative stress may result from direct ROS formation via autooxidation in the presence of transition metals, activation of oxidant systems, and inhibition of antioxidant systems [2][3]. Impairment of epigenetic control mechanisms of gene expression, such as DNA methylation, histone modification, and non-coding RNA, is another possible mechanism of Hcy toxicity [4]. Beyond this, Hcy can change structure and function of proteins by binding to their lysine or cysteine residues; these post-translational modifications (PTMs) are known as N- and S-homocysteinylation, respectively. These mechanisms of Hcy-mediated injury are not mutually exclusive, since altered expression and PTMs of proteins involved in prooxidant/antioxidant pathways can lead to increased cellular oxidative stress and, oppositely, free radicals can induce alterations in gene expression and oxidative PTMs of proteins.
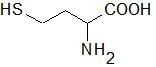
Figure 1. Structural formula of homocysteine (Mr = 135.2).
Mitochondria fulfill many essential cellular functions, and mitochondrial disorders are associated with development of various neurological and cardiovascular diseases. Hcy, a widely accepted risk factor for these diseases, affects normal mitochondrial structure and function, including energy production, mitochondrial dynamics, and cell survival and death (Figure 2).
2. Mitochondrial Energy Metabolism and Oxidative Stress
Mitochondria play an essential role in energy production via the electron transport chain (ETC) coupled with oxidative phosphorylation (OXPHOS). The series of redox reactions provide free energy, which drives pumping of protons into the intermembrane space creating an H+ electrochemical gradient–membrane potential (Ψm) across the inner mitochondrial membrane. In turn, this membrane potential is used as a source of energy to synthesize ATP by complex ATP synthase. HHcy results in decreased mitochondrial respiration associated with reduced activities of ETC complexes and diminished ATP production [5][6][7][8]. Moreover, ETC seems to be a major source of ROS under HHcy [6][9]. ROS-induced damage to mitochondrial DNA and proteins was demonstrated in the cardiac, brain, and vascular tissues [6][7][10]. HHcy also affects expression of enzyme antioxidants via Keap1-Nrf2 signaling pathway, the major regulator of cellular redox homeostasis. Diverse mechanisms through which Hcy can modulate this pathway under different conditions may result in activation or inhibition of antioxidant enzymes [11][12][13].
3. Homocysteine and Mitochondrial Apoptotic Pathway
ROS and mitochondrial damage promote the intrinsic, also called mitochondrial, apoptotic pathway. Decreases in membrane potential and ATP content and increased mitochondrial membrane permeability allow for translocation of pro-apoptic factors leading to the activation of the caspase signaling pathway and apoptosis. In cardiovascular and cerebrovascular systems, HHcy induces apoptosis via disturbed mitochondrial homeostasis and oxidative stress. Decreased mitochondrial membrane potential, upregulation of pro-apoptotic and downregulation of anti-apoptotic proteins, release of cytochrome c, or activation of caspase-9 and its downstream caspase-3 was shown in different cell lines, as well as in animal models [9][12][14].
Inflammation plays an important role in vascular disease and hypertension. HHcy promotes vascular inflammation via activation of Toll-like receptor 4 (TLR-4), leading to initiation of mitochondrial apoptotic cell death [14].
4. Homocysteine and Mitochondrial Dynamics
Mitochondrial dynamics is an important mechanism of regulation of mitochondrial morphology and function. This term encompasses mitochondrial fusion, fission, transportation, and selective degradation—mitophagy. In mammalian cells, mitochondrial fusion is controlled by mitofusins 1 and 2 (Mfn 1 and Mfn2, respectively) located in the outer mitochondrial membrane, and optic atrophy 1 (Opa1) protein, which regulates fusion of inner membranes. Mitochondrial fission is mediated by cytosolic GTPase dynamin-related protein (DRP1), which binds to outer membrane receptors, like Fis 1, to constrict both outer and inner membranes and divide the mitochondrion into two daughter organelles. By fission, a daughter mitochondrion, which may contain deleterious or damaged components, is removed via mitophagy. In mouse brain endothelial cells, Hcy upregulated DRP1, Mfn2, and autophagy marker LC-3 [15]. Studies on retinal ganglion neurons show Hcy-induced disbalance of mitochondrial fusion and fission. Using cystathione-β-synthase-deficient mice as a model of moderate HHcy, increased expression of Opa 1 and Fis 1 proteins in retinal ganglion neurons was demonstrated [16]. The alterations in Opa 1 and Fis 1 levels were associated with increased mitochondrial fission as suggested by the increased number of small mitochondria [16].
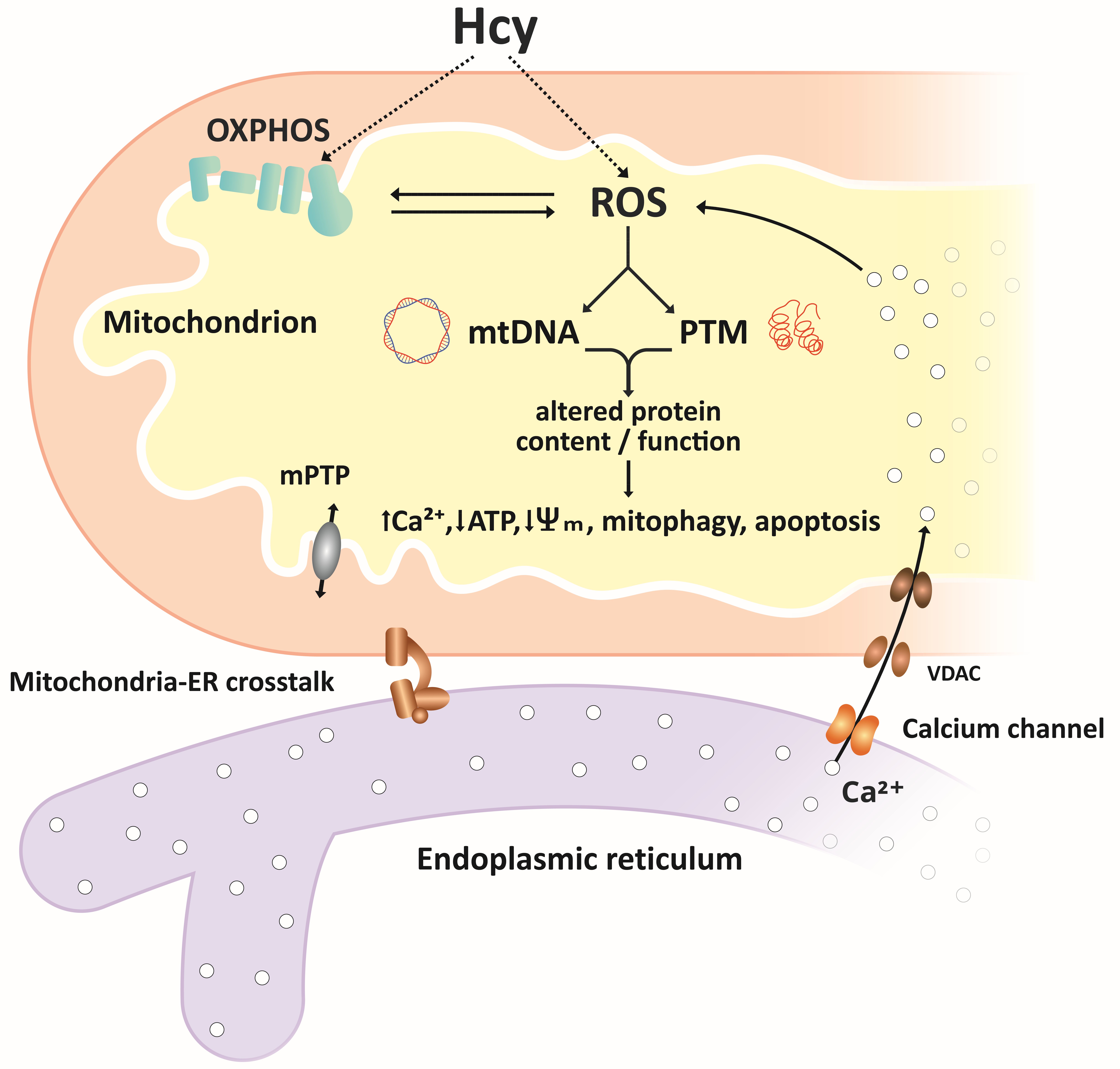
Figure 2. Simplified overview of homocysteine effects on mitochondria. Reactive oxygen species ROS, produced in the electron transport chain ETC or through autooxidation of Hcy, are important mediators of Hcy effects on mitochondrial function. ROS-associated oxidative damage to mtDNA and post-translational modifications PTMs of proteins result in altered content and/or function of mitochondrial proteins, including components of the ETC, antioxidant/pro-oxidant enzymes, membrane carriers, and receptors. Altered function of mitochondrial proteins may result in further increases in ROS levels, accumulation of Ca2+ ions mediated by mitochondria–ER crosstalk, decrease in ATP production, decreased membrane potential, and initiation of mitophagy and apoptosis.
References
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidatice stress. Amino Acids 2003, 25, 409–417.
- Perna, A.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; De Santo, N.G. Possible mechanisms of homocysteine toxicity. Kidney Int. 2003, 63, S137–S140.
- Esse, R.; Barroso, M.; De Almeida, I.T.; Castro, R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state-of-the-art. J. Mol. Sci. 2019, 20, 867.
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of epigenetic mechanisms of gene expression in the pathologies of hyperhomocysteinemia. J. Mol. Sci. 2019, 20, 3140.
- Timkova, V.; Tatarkova, Z.; Lehotský, J.; Racay, P.; Dobrota, D.; Kaplán, P. Effects of mild hyperhomocysteinemia on electron transport chain complexes, oxidative stress, and protein expression in rat cardiac mitochondria. Cell. Biochem. 2016, 411, 261–270.
- Longoni, A.; Kolling, J.; Siebert, C.; Dos Santos, J.P.; Da Silva, J.S.; Pettenuzzo, L.F.; Meira-Martins, L.A.; Gonçalves, C.-A.; De Assis, A.M.; Wyse, A.T.S. 1,25-Dihydroxyvitamin D 3 prevents deleterious effects of homocysteine on mitochondrial function and redox status in heart slices. Res. 2017, 38, 52–63.
- Folbergrová, J.; Ješina, P.; Drahota, Z.; Lisý, V.; Haugvicová, R.; Pecinová, A.; Houstek, J. Mitochondrial˘ complex I inhibition in cerebral cortex of immature rats following homocysteic acid-induced seizures. Neurol. 2007, 204, 597–609.
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced mitochondrial dysfunctions in brain. Mitochondrion 2020, 50, 158–169.
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Sun, B.L.; Fu, X.Y. Astaxanthin attenuates homocysteine-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage. Physiol. 2017, 8, 1041.
- Chou, Y.-F.; Yu, C.-C.; Huang, R.-F.S. Changes in mitochondrial DNA deletion, content, and biogenesis in folate-deficient tissues of young rats depend on mitochondrial folate and oxidative DNA injuries. Nutr. 2007, 137, 2036–2042.
- Kumar, M.; Sandhir, R. Neuroprotective efect of hydrogen sulfide in hyperhomocysteinemia is mediated through antioxidant action involving Nrf2. Med. 2018, 20, 475–490.
- Wu, B.; Yue, H.; Zhou, G.H.; Zhu, Y.Y.; Wu, T.H.; Wen, J.F.; Cho, K.W.; Jin, S.N. Protective effects of oxymatrine on homocysteine-induced endothelial injury: Involvement of mitochondria-dependent apoptosis and Akt-eNOS-NO signaling pathways. J. Pharmacol. 2019, 864, 172717.
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.-C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59.
- Familtseva, A.; Chaturvedi, P.; Kalani, A.; Jeremic, N.; Metreveli, N.; Kunkel, G.H.; Tyagi, S.C. Toll-like receptor 4 mutation suppresses hyperhomocysteinemia-induced hypertension. J. Physiol. Cell. Physiol. 2016, 311, C596–C606.
- Vacek, J.C.; Behera, J.; George, A.K.; Kamat, P.K.; Kalani, A.; Tyagi, N. Tetrahydrocurcumin ameliorates homocysteine-mediated mitochondrial remodeling in brain endothelial cells. Cell. Physiol. 2017, 233, 3080–3092.
- Ganapathy, P.S.; Perry, R.L.; Tawfik, A.; Smith, R.M.; Perry, E.; Roon, P.; Bozard, B.R.; Ha, Y.; Smith, S.B. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Investig. Opthalmol. Vis. Sci. 2011, 52, 5551–5558.

