 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Philippe Le Mercier | + 2193 word(s) | 2193 | 2020-11-02 10:55:58 | | | |
| 2 | Philippe Le Mercier | Meta information modification | 2193 | 2020-11-09 16:09:48 | | | | |
| 3 | Rita Xu | -441 word(s) | 1752 | 2020-11-10 05:18:05 | | |
Video Upload Options
Virosaurus database offers curated virus sequences, available at various degree of clustering. Clustering virus sequences with different similarity scores gives an indication of the genetic diversity of a each virus, and how deep it is.
For example HIV-1 and Influenza sequences present high numbers of clusters when clustered at 98% similarity. At 90% similarity, the number of influenza virus clusters dramatically drops by a factor of about 20, when for HIV-1it drops by a factor of 2. This suggest that the diversity of HIV-1 sequence is somehow deeper that the one for influenza.
1. Introduction
At least 130 different viruses infecting humans have been recorded so far [1]. Diagnostic assays rely on specific procedures for each virus screened, mostly by using enzyme-linked immunosorbent assay (ELISA) or molecular techniques such as real-time polymerase chain reaction (qPCR) [2]. These tests are robust but are, by design, restricted to what the researcher is looking for. Moreover, some viruses resist diagnostics by these methods because of their unusual genetic diversity, such as Lassa virus [3]. Classical diagnosis methods are adapted in routine diagnosis, but show limitations for some research, such as viral surveillance or virus discovery investigations that require testing all known viruses in one sample [4][5].
High-Throughput Sequencing (HTS) technology makes possible the detection of all genetic entities present in a sample. Thus, this method offers promising perspectives for virus detection and diagnosis [6]. HTS produces millions of reads that are identified by comparison with a virus reference dataset. Many laboratories have set up their HTS virus diagnostic pipeline by building up their own reference database, mostly by cherry-picking virus reference sequences from NCBI/GenBank or UniProt resources [7][8][9][10]. Building a consistent reference database is a challenge because viruses are extremely diverse and variable [11][12]. Creating a non-biased virus reference database requires comparing all viral genomic material to identify the minimum number of sequences needed to cover all genetic diversity at a given percentage of identity [13].
To fill these requirements, Virosaurus (for Virus Thesaurus) is an unbiased viral database built from eukaryotic virus genomes publicly available, cleaned and annotated. The focus of this database is not only to provide accurate references of virus diversity, but also to add a layer of annotation for better clinical analysis of results. Other virus reference databases have been published but with a different scope or annotations. VirusSITE is a virus reference database published in 2016 [14], mainly based on NCBI RefSeq sequences [15]. In 2018, Arifa Khan’s group at the Food and Drug Administration (FDA) released the Reference Viral Database (RVDB) for virus detection using high-throughput sequencing [16], which is also based on RefSeq [15], and enlarged by a similarity search. Virosaurus has a different approach, being directly built on all GenBank sequences with the addition of a layer of clinical annotation.
2. Clinical Annotation
The complete virus sequence dataset has been annotated by adding controlled vocabulary metadata in the FASTA files (Figure 1). All sequence taxonomy has been normalized to the species level, thereby matching reads can be classified under one viral entity (Figure 2).
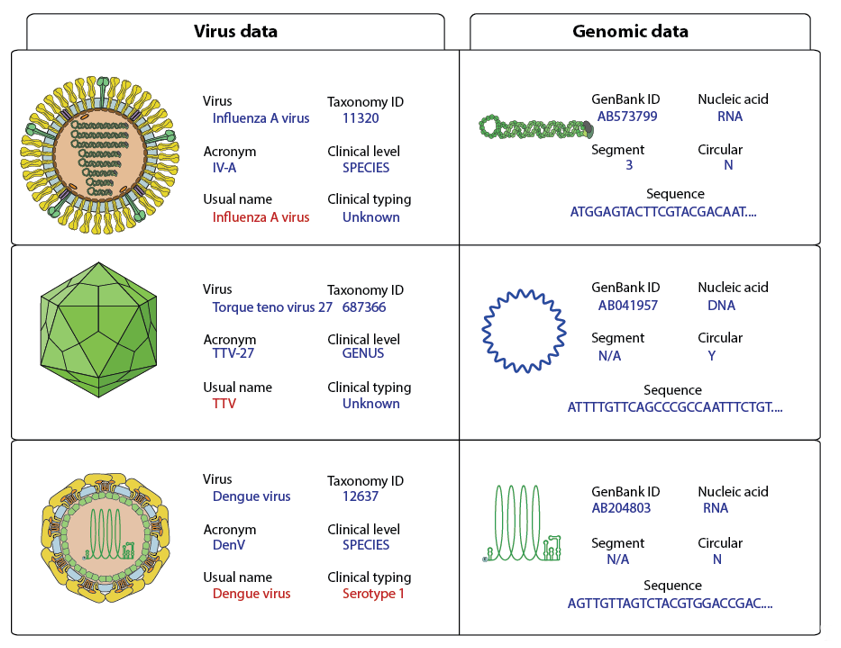
Figure 1. Examples of virus genome annotation. The usual name and clinical typing should be the default output for clinical studies and are shown in red.
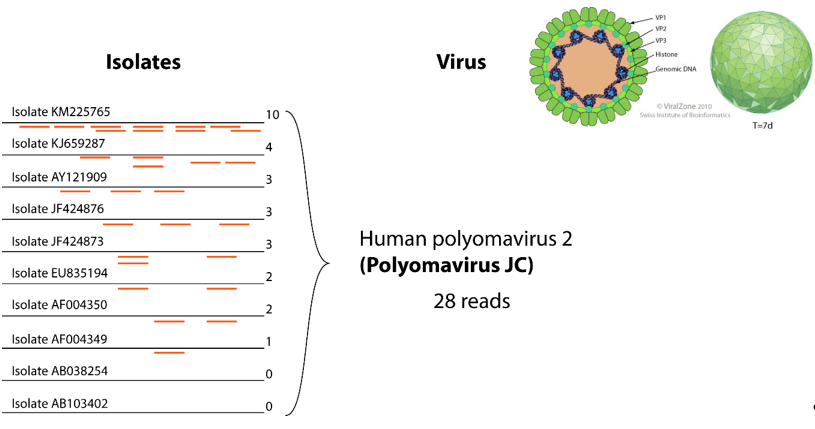
Figure 2. Example of gathering reads for the same virus. In the left part, 10 isolates represent clusters for this virus. Twenty-eight reads show homology to those reference sequences, they can be all grouped under the “human polyomavirus 2” entity, thereby facilitating interpretation of results.
Metadata about the viral genome comprises GenBank accession number, nucleic acid nature, number of segments, circular/linear and sequence. Considering that some sample preparation protocols target either RNA or DNA, it is essential to know if the viral genome is DNA or RNA for virus detection. For example, RNA virus detection in DNA preparations should trigger further investigations to rule-out contaminants. Whether the genome is circular or linear matters if users want to assemble HTS reads. In the case of circular genomes, reads can overlap at the start and end of the sequence. All metadata are added to the FASTA files in the header section of each sequence (Figure 1).
Each sequence in the INSDC database is linked to a virus isolate. Nonetheless, isolate data are not useful for clinicians in daily routine. To propose a relevant output, the Virosaurus database was designed to offer a broader classification of viruses at a higher level (Figure 2). We have used taxonomic classification: all sequences have been classified at the species level and annotation has been added about clinical virus belonging to this taxa: species name, TaxonomyID, acronym, usual name, clinical level and typing. The “usual name” displays the common virus name, which can be easier to interpret and more stable than the official name. For example, parvovirus B19 is the common name of a virus belonging to Primate erythroparvovirus 1 [17], most people are more familiar with the first than the latter name. Moreover, some viruses classification is not directly relevant to a clinical interest: there are 29 torque teno virus named Torque teno virus 1 to 29 and several of them can infect the same person [18]. Having a list of TTV<numbers> was not relevant for clinicians: the entire Torque teno virus received the common name “TTV”. The same has been done for alphapapillomaviruses, which have all “HPV” as “usual name”. These exceptions are signified in the field “clinical level”, describing the taxonomy level for which these names have been assigned to sequences. The annotated field “clinical typing” comprises genotype (ex: HCV; Norwalk virus), serotype (ex: Dengue virus) or disease (ex: polio, High risk HPV) when these data are necessary for some viruses.
4. Clustering
Because virus genomes are small and thus relatively easy to sequence, thousands of genome sequences are available, and these datasets are highly redundant. Redundancy can result in bias, and increases computational time with no benefits. Clustering computer programs are designed to removing redundancy by alignment methods. This consists of calculating clusters of similar sequences at a given percentage of similarity, and selecting one sequence to represent the cluster. We used CD-HIT to cluster our complete virus sequence dataset at 90% and 98% similarity thereby creating Virosaurus90 and Virosaurus98 [19]. Clusters were then reviewed to validate the quality of data. Ideally, each cluster should contain only sequences belonging to a single species. Clusters comprising sequences from more than one species were rare and manually checked. This low intermixing underlines the quality of taxonomic classification by ICTV [20].
Figure 3 displays data about the 13 most sequenced human viruses in GenBank. Interestingly, the relative composition of the datasets changes drastically between complete virus sequences, Virosaurus98 and 90 (Figure 3C,D). Influenza virus sequences are reduced 25 times by clustering at 98%, revealing a high redundancy, whereas HIV-1 sequences are reduced 1.3 times. Clustering at 90% reduces the number of influenza sequences 760 times, whereas HIV-1 sequences are reduced 2.8 times. This indicates that we have highly redundant sequences of influenza viruses in which the diversity drops at 90% similarity. In contrast, HIV-1 complete genomes are less redundant, but with a higher diversity. HIV recombinants might play a role in this observed diversity [21] as CD-HIT aligns full genomes and will create new clusters for most recombinants.
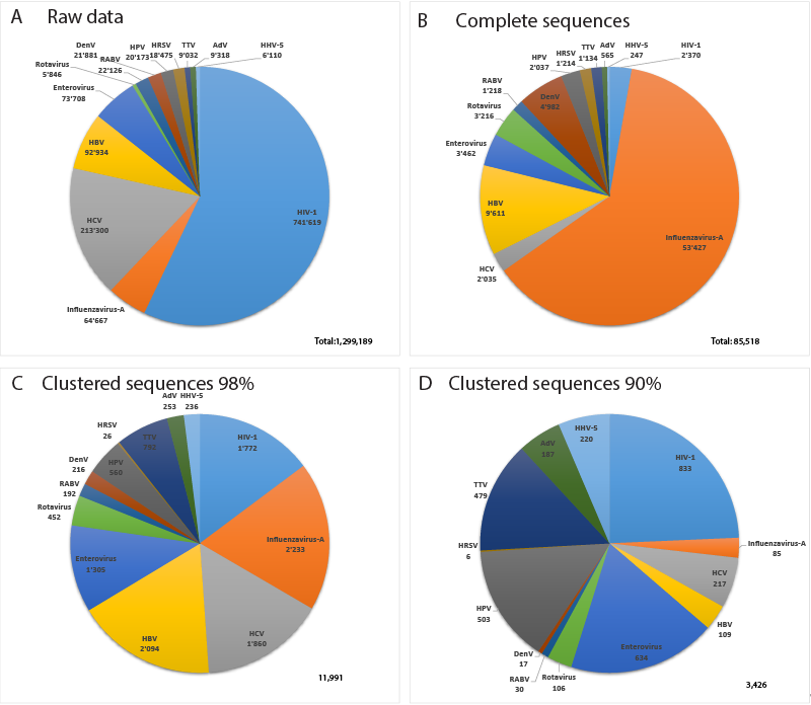
Figure 3. Relative number of sequences for the 13 most sequenced human viruses: (A) total sequences from GenBank, (B) complete virus sequences, (C) Virosaurus 98 and (D) Virosaurus 90. (Data from release 2019_10).
5. Reducing False Negatives in Virus Detection
Detection of viruses by HTS and bioinformatics analysis depends on the quality of the references. If the latter is suboptimal, this could lead to false negatives in the detection results for important viruses. Do we have enough knowledge of each viruses’ genetics to detect circulating viruses? The clustering method allows estimation of the genetic variability of a virus. A virus for which the genetic landscape has been thoroughly mapped will comprise many sequences in each cluster. On the other hand, we have only partial knowledge when clusters are isolated and contain few or a single sequence. Figure 4 shows the percent reduction of sequences by clustering at 90%. A low percentage suggests that we do not have significant data about the genetic landscape of a given virus. Some viruses like Influenza A virus, Dengue virus, Zika virus or ebolaviruses display a reduction of over 98%, meaning that we have at our disposal many more sequences than we probably need to detect those viruses in HTS samples. Of course, new strains may emerge but at least we have sampled much of the prior genetic data. Herpesviruses and poxviruses are the only viruses displaying a reduction inferior to 50%. In other words, the majority of clusters contain a single sequence. The worst cases are Human betaherpesvirus 6A and 6B for which clustering does not reduce the number of sequences, demonstrating a complete absence of redundancy in the current databases.
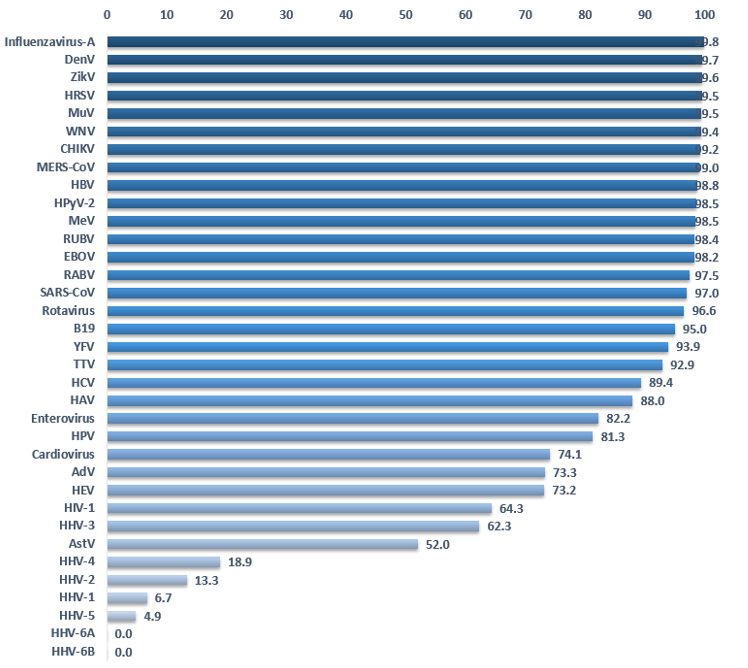
Figure 4. Percentage of sequence reduction by clustering complete genomes at 90%. (Data Virosaurus 2019_10).
This lack of redundancy suggests that herpesviruses and poxviruses are more diverse than we know, and we might be unable to detect all circulating viruses with our reference dataset. We decided to use a different method to gather sequences for these two virus families. Since there are too few complete genomes, we have used genes instead of genomes (Figure 1). Using genes instead of genomes allows accessing many more sequences and maximizes the chances of identifying a virus with the reference dataset. Because the objective of Virosaurus is to analyze genetic entities at the level of species, using individual genes does not change the output post-processing.
6. Validation on Clinical Samples
The Virosaurus database 2018_11 has been tested on sequencing data from 20 human clinical samples previously characterized by HTS using the ezVIR pipeline (Figure 5) [9][17][18][20][22][23][21][24]. The results of this comparative analysis show 100% concordance. Moreover, the database has been used in a pilot study to characterize viruses causing fever, and provides a surveillance tool for emerging viral diseases in Gabon [25]. The results are concordant with the PCR screening results from a previous study [26].
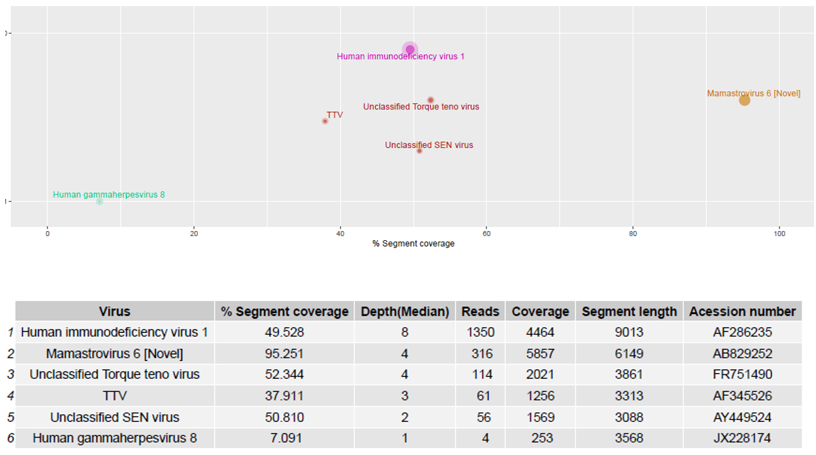
Figure 5. Human blood samples were sequenced and reads generated using the RNA protocol [9] were aligned to the Virosaurus database. The result is easy to interpret and confirms that the patient was positive for a novel human astrovirus, HIV-1 and HHV-8 sequences, as previously reported [20]. Top panel: 2D representation of detected sequences with %segment coverage in the X-axis, and depth (median) in Y-axis; bottom panel: raw data. Size of dots is relative to number of reads. Anellovirus (TTV) sequences were also detected. The Virosaurus hierarchy allows allocating reads to viral entities: at the level of virus (HIV-1, HHV-8, MastV-6) or higher (TTV). Unclassified SEN viruses are TTV-like genomes. Mamastrovirus (Novel) is a subtyping, allowing differentiating between novel (i.e., MLB and VA/HMO) and classical human astroviruses.
References
- Human viruses table—ViralZone page. Available online: https://swissprot.sib.swiss/prime/678 (accessed on 9 July 2020).
- Diagnostic Methods in Virology, Virological Methods, Virus Culture, Virus Isolation. Available online: http://virology-online.com/general/Test1.htm (accessed on 9 July 2020).
- Happi, A.N.; Happi, C.T.; Schoepp, R.J. Lassa fever diagnostics: Past, present, and future. Curr. Opin. Virol. 2019, 37, 132–138, doi:1016/j.coviro.2019.08.002.
- Lipkin, W.I.; Firth, C. Viral surveillance and discovery. Opin. Virol. 2013, 3, 199–204, doi:10.1016/j.coviro.2013.03.010.
- Lipkin, W.I.; Anthony, S.J. Virus hunting. Virology 2015, 479–480, 194–199, doi:1016/j.virol.2015.02.006.
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Rev. Genet. 2019, 20, 341–355, doi:10.1038/s41576-019-0113-7.
- Lewandowska, D.W.; Zagordi, O.; Zbinden, A.; Schuurmans, M.M.; Schreiber, P.; Geissberger, F.-D.; Huder, J.B.; Böni, J.; Benden, C.; Mueller, N.J.; et al. Unbiased metagenomic sequencing complements specific routine diagnostic methods and increases chances to detect rare viral strains. Microbiol. Infect. Dis. 2015, 83, 133–138, doi:10.1016/j.diagmicrobio.2015.06.017.
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Microbiol 2018, 9, 749, doi:10.3389/fmicb.2018.00749.
- Petty, T.J.; Cordey, S.; Padioleau, I.; Docquier, M.; Turin, L.; Preynat-Seauve, O.; Zdobnov, E.M.; Kaiser, L. Comprehensive human virus screening using high-throughput sequencing with a user-friendly representation of bioinformatics analysis: A pilot study. Clin. Microbiol. 2014, 52, 3351–3361, doi:10.1128/JCM.01389-14.
- Xu, H.; Ling, Y.; Xi, Y.; Ma, H.; Wang, H.; Hu, H.-M.; Liu, Q.; Li, Y.-M.; Deng, X.-T.; Yang, S.-X.; et al. Viral metagenomics updated the prevalence of human papillomavirus types in anogenital warts. Emerg. Microbes 2019, 8, 1291–1299, doi:10.1080/22221751.2019.1661757.
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142, doi:1016/j.virusres.2017.02.002.
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2, vew022, doi:1093/ve/vew022.
- Kiselev, D.; Matsvay, A.; Abramov, I.; Dedkov, V.; Shipulin, G.; Khafizov, K. Current Trends in Diagnostics of Viral Infections of Unknown Etiology. Viruses 2020, 12, doi:3390/v12020211.
- Stano, M.; Beke, G.; Klucar, L. viruSITE-integrated database for viral genomics. Database (Oxford) 2016, 2016, doi:1093/database/baw162.
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D7337–D7345, doi:1093/nar/gkv1189.
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, doi:10.1128/mSphereDirect.00069-18.
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247.
- Ball, J.K.; Curran, R.; Berridge, S.; Grabowska, A.M.; Jameson, C.L.; Thomson, B.J.; Irving, W.L.; Sharp, P.M. TT virus sequence heterogeneity in vivo: Evidence for co-infection with multiple genetic types. J. Gen. Virol. 1999, 80, 1759–1768.
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152.
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717.
- Vuilleumier, S.; Bonhoeffer, S. Contribution of recombination to the evolutionary history of HIV. Curr. Opin. HIV AIDS 2015, 10, 84–89.
- Cordey, S.; Hartley, M.-A.; Keitel, K.; Laubscher, F.; Brito, F.; Junier, T.; Kagoro, F.; Samaka, J.; Masimba, J.; Said, Z.; et al. Detection of novel astroviruses MLB1 and MLB2 in the sera of febrile Tanzanian children. Emerg. Microbes Infect. 2018, 7, 27.
- Williams, S.H.; Cordey, S.; Bhuva, N.; Laubscher, F.; Hartley, M.-A.; Boillat-Blanco, N.; Mbarack, Z.; Samaka, J.; Mlaganile, T.; Jain, K.; et al. Investigation of the Plasma Virome from Cases of Unexplained Febrile Illness in Tanzania from 2013 to 2014: A Comparative Analysis between Unbiased and VirCapSeq-VERT High-Throughput Sequencing Approaches. mSphere 2018, 3.
- Cordey, S.; Laubscher, F.; Hartley, M.-A.; Junier, T.; Pérez-Rodriguez, F.J.; Keitel, K.; Vieille, G.; Samaka, J.; Mlaganile, T.; Kagoro, F.; et al. Detection of dicistroviruses RNA in blood of febrile Tanzanian children. Emerg. Microbes Infect. 2019, 8, 613–623.
- Fernandes, J.F.; Laubscher, F.; Held, J.; Eckerle, I.; Docquier, M.; Grobusch, M.P.; Mordmüller, B.; Kaiser, L.; Cordey, S. Unbiased metagenomic next-generation sequencing of blood from hospitalized febrile children in Gabon. Emerg. Microbes Infect. 2020, 9, 1242–1244.
- Fernandes, J.F.; Held, J.; Dorn, M.; Lalremruata, A.; Schaumburg, F.; Alabi, A.; Agbanrin, M.D.; Kokou, C.; Ben Adande, A.; Esen, M.; et al. Causes of fever in Gabonese children: A cross-sectional hospital-based study. Sci Rep. 2020, 10, 2080.



