 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yuh-Cherng Chai | + 3048 word(s) | 3048 | 2020-11-05 05:14:40 | | | |
| 2 | Yuh-Cherng Chai | -28 word(s) | 3020 | 2020-11-09 00:26:30 | | | | |
| 3 | Yuh-Cherng Chai | -28 word(s) | 3020 | 2020-11-09 00:36:42 | | | | |
| 4 | Yuh-Cherng Chai | -28 word(s) | 3020 | 2020-11-09 00:44:48 | | | | |
| 5 | Catherine Yang | Meta information modification | 3020 | 2020-11-10 04:10:51 | | |
Video Upload Options
S-glutathionylation, the post-translational modification forming mixed disulfides between protein reactive thiols and glutathione, regulates redox-based signaling events in the cell and serves as a protective mechanism against oxidative damage. S-glutathionylation alters protein function, interactions, and localization across physiological processes, and its aberrant function is implicated in various human diseases.
1. Introduction
In mammalian cells, S-glutathionylation can function as a regulatory mechanism or protect proteins against irreversible oxidation. By introducing the tripeptide glutathione with additional ionic charges into a protein, S-glutathionylation resembles the well-characterized mechanism of protein phosphorylation. The dynamic and reversible nature of S-glutathionylation highlights its ability to function as a redox “switch”, regulating protein function, interactions, and localization[1].
2. Molecular Mechanisms of Protein S-Glutathionylation
The modification of protein-reactive thiols with glutathione was initially thought to be a non-specific reaction induced in pathological, highly oxidative situations or upon exposure to strong oxidants, particularly under in vitro conditions[2]. Recent studies, however, have demonstrated that protein S-glutathionylation is a dynamic process that may also occur under physiological conditions. A number of chemical mechanisms by which protein thiols can interact with the cellular glutathione pool, leading to their S-glutathionylation have been proposed. Figure 1 shows a summary of the potential mechanisms of S-glutathionylation that are discussed in more detail below.
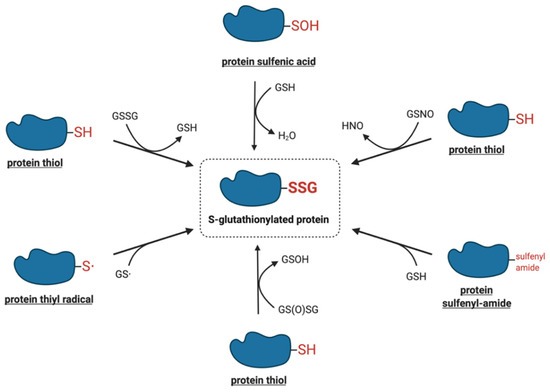
Figure 1. Proposed molecular mechanisms of S-glutathionylation.
2.1. Thiol-Disulfide Exchange Mechanism
One of the most intensively studied mechanisms for protein S-glutathionylation involves a thiol-disulfide exchange reaction[3]. The thiol-disulfide exchange mechanism primarily relies on the redox state of cellular glutathione. In principle, oxidative stress could result in the oxidation of GSH molecules to form GSSG, shifting the GSH:GSSG redox balance toward a more oxidizing state. As a result, it was hypothesized that the accumulation of GSSG would lead to a spontaneous thiol-disulfide exchange between protein cysteine thiols and GSSG, thereby yielding the corresponding protein-mixed disulfide along with GSH (Equation (1)). In this mechanism, the extent of protein S-glutathionylation ([PSSG]:[PSH]) is dependent on the intracellular GSH:GSSG ratio. Therefore, the equilibrium constant of the reaction (Kmix, Equation (2)) represents the specific oxidation potential for the formation of the mixed disulfide.
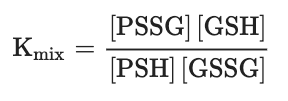 (Equation 2)
(Equation 2)For most protein cysteines, Kmix is approximately 1, indicating that the GSH:GSSG ratio would have to decline dramatically to drive S-glutathionylation[3]. The ratio of GSH:GSSG, however, usually remains very high inside the cytosol, even during extreme conditions of oxidative stress. This is due to the capability of most cells to increase the activity of glutathione reductase or actively transport GSSG out of the cell as a protective mechanism against oxidative stress[4]. Thus, although the thiol–disulfide exchange of a protein thiolate with GSSG may occur spontaneously, the reaction is slow due to a lack of extreme conditions. The absence of a detectable increase in GSSG during S-glutathionylation stimulated by the respiratory burst in human neutrophils provides evidence against a thiol–disulfide exchange mechanism[5]. Therefore, except for proteins, such as the transcription factor c-Jun, that display unusually high thiol redox potentials (Kmix~13)[6], the thiol–disulfide exchange reaction is not a likely mechanism for S-glutathionylation, and proteins shown to be S-glutathionylated by GSSG in vitro may not reflect what occurs in vivo.
2.2 Reactive Thiol Intermediates for S-Glutathionylation
The mechanisms that are more likely to mediate S-glutathionylation reactions in vivo involve reactive thiol derivates such as sulfenic acids, sulfenyl-amides, thiyl radicals, S-nitrosylated thiols, and thiosulfinates.
2.2.1. Sulfenic Acids
The two-electron oxidation of a cysteine thiolate generates a sulfenic acid, the simplest oxyacid of sulfur. The oxidants most commonly implicated in the conversion of protein thiolates to their corresponding sulfenic acids include hydrogen peroxide, alkyl hydroperoxides, peroxynitrite, hypochlorous acid, and chloramines[7]. Under prolonged exposure to these oxidants, sulfenic acids may be oxidized to more stable and irreversible thiol derivatives such as sulfinic and sulfonic acids or may react with neighboring thiols to form disulfides[7]. The abundance of GSH in cells is known to rapidly react with protein sulfenic acids to displace the hydroxyl group and form S-glutathionylated proteins[7]. It is plausible that a glutathione sulfenic acid could react by a similar mechanism with a protein thiolate to generate the S-glutathionylated protein, but the low acidity of the GSH thiol (pKa = 9.6) is likely to limit this reaction in vivo[8].
Many proteins have been identified as candidates for regulation by sulfenic acid formation. However, most of these studies were performed in vitro and in the absence of GSH. Recent literature suggests that although sulfenic acid formation may be the initial oxidative modification for these proteins, S-glutathionylation serves as the more stable intermediate in redox signaling[9]. This was shown for the molecular chaperone BiP, initially suggested to be regulated by the sulfenic acid formation and later shown to undergo S-glutathionylation through a sulfenic acid intermediate[10]. Similarly, mammalian protein tyrosine phosphatases (PTPs), initially observed to react with H2O2 to form a protein sulfenic acid[11][12], have been shown to undergo S-glutathionylation in the presence of GSH[13]. More recently, Heppner et al. showed that the oxidation of epidermal growth factor receptor (EGFR) and non-receptor tyrosine kinase Src by the NADPH oxidase DUOX1 involved sequential oxidation to sulfenic acids and S-glutathionylated proteins[14]. In agreement with these findings, sulfenic acids have been shown to react much faster with GSH than with oxidants like H2O2, thereby escaping further oxidative modification[15].
2.2.2. Sulfenyl-Amides
The formation of the sulfenyl-amide species was first demonstrated in vitro by Salmeen et al., as an alternative protective modification for the active site cysteine of protein tyrosine phosphatase 1B (PTP1B)[16]. The proposed mechanism involved the initial oxidation of Cys215 via H2O2 to sulfenic acid, followed by the rapid elimination of oxygen to generate a more stable derivative, the sulfenyl-amide species[16]. X-ray crystallographic analysis showed that the sulfur atom of Cys215 formed a cyclic covalent bond to the primary chain nitrogen of an adjacent residue, resulting in a conformational change in the catalytic site of the protein[16]. The sulfenyl-amide bond was fully reducible upon the addition of GSH. As earlier studies had demonstrated that PTP1B activity could be regulated by the S-glutathionylation of Cys215[14], the sulfenyl-amide bond was proposed to serve as an intermediate to facilitate the formation of S-glutathionylated PTP1B[16]. At present, whether this mechanism is relevant in vivo or for proteins other than PTP1B remains unknown.
2.2.3. Thiyl Radicals
Thiyl radicals are highly reactive species that can be generated via electron transfer or hydrogen atom abstraction from a thiol[17][18]. Free radical species that oxidize thiols to thiyl radicals include hydroxyl (•OH), carbonate (CO3•–), nitrogen dioxide (•NO2), superoxide (O2•–), and peroxyl and phenoxyl radicals[19].
The formation of protein thiyl radicals or glutathione thiyl radicals may lead to protein S-glutathionylation reactions through radical recombination or reaction of a radical with a thiolate followed by reaction with O2 [9]. In the presence of glutathione thiyl radical-generating systems (Fe2+/ADP/H2O2 + GSH or horseradish peroxidase/H2O2 + GSH), several proteins have been shown to undergo S-glutathionylation in vitro[20]. A recent study by Kang et al., shows that a burst in the mitochondrial superoxide production leads to thiyl radical formation resulting in S-glutathionylation of the mitochondrial Complex I[21][22]. Other researchers have discovered that superoxide induces endothelial nitric-oxide synthase (eNOS) protein thiyl radical formation, leading to the modification of cysteine with either disulfide bond formation or S-glutathionylation[23][24]. Thus, it seems likely that thiyl radicals may serve as regulatory intermediates leading to S-glutathionylation reactions in vivo. Moreover, there is evidence that these reactions may be catalyzed by glutaredoxin[9].
2.2.4. S-Nitrosylated Thiols
S-nitrosylation is a type of post-translational modification that can occur under normal and pathological cellular conditions, analogous to S-glutathionylation [25]. It is characterized by the coupling of a nitric oxide (·NO) moiety to a reactive cysteine thiol, forming an S-nitrosothiol[26][27]. The S-nitrosothiol of glutathione (GSNO) is a relatively stable molecule found in micromolar concentrations in healthy tissues[9]. Although a variety of reagents, including nitrous acid, nitrogen dioxide and nitrosyl chloride can react with GSH to induce GSNO formation, dinitrogen trioxide (N2O3) is considered to be the primary nitrosylating agent[28]. During conditions of nitrosative stress, specific cell compartments can produce higher levels of GSNO, which can promote both the S-nitrosylation and S-glutathionylation of proteins[29][30]. Alternatively, the reaction between a protein S-nitrosothiol and glutathione may also lead to S-glutathionylation, although there is little information regarding this mechanism.
In the presence of physiologically relevant concentrations of GSH, one study showed that NO inhibits the DNA binding activity of the transcription factor c-Jun by specifically inducing S-glutathionylation in its DNA binding site[31]. Another study with isolated proteins showed that papain, creatine phosphokinase, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were significantly both S-nitrosylated and S-glutathionylated by GSNO, whereas alcohol dehydrogenase, bovine serum albumin, and actin were only S-nitrosylated[32]. The S-nitrosylation of GAPDH was shown to be unstable and decompose spontaneously, whereas S-glutathionylation led to the inhibition of the enzyme[33]. Thus, it is plausible that S-nitrosylation and S-glutathionylation may exert opposing effects on protein function[34]. At present, the structural features that select S-glutathionylation over S-nitrosylation or vice versa remain largely uncertain but are likely related to the local microenvironment of the target cysteine.
2.2.5. Thiosulfinates
Glutathione thiosulfinate (also called glutathione disulfide S-oxide, GS(O)SG) is the anhydride of glutathione sulfenic acid[35]. Li et al. identified this unique molecule as a product derived from the spontaneous decomposition of GSNO in aqueous solution. The mechanism for the generation of glutathione thiosulfinate is not yet known, although it has been speculated that the initiation of GSNO decomposition may be catalyzed by copper ions through homolysis, yielding GSH and NO[35][28]. Glutathione thiosulfinate may also be generated through the oxidation of a disulfide or thiol by the O2•–/H2O2-producing system xanthine/xanthine oxidase[35][36].
Glutathione thiosulfinate is highly reactive with thiols and has been suggested to mediate S-glutathionylation of several proteins. In the rat brain, glutathione thiosulfinate was shown to be one of the most potent S-glutathionylating agent among the various glutathione derivatives tested, including GSNO and GSSG[36]. As glutathione thiosulfinate is the degradation product of GSNO, its formation may account for some of the effects of GSNO-induced S-glutathionylation[36]. The formation of the thiosulfinate product has been proposed to mediate the S-glutathionylation of rat brain neurogranin (Ng)[36], matrix metalloproteinases[37], and tyrosine hydroxylase[38]. At present, it remains unclear whether glutathione thiosulfinate or other glutathione derivatives (sulfenic acid, thiyl radical, etc.) play more critical roles as intermediates in the S-glutathionylation mechanism in vivo.
3. Enzymatic Protein S-Glutathionylation
Although protein S-glutathionylation in cells may involve non-enzymatic chemical reactions, several enzymes have been proposed to catalyze the transfer of glutathione to the target protein.
3.1. Glutathione S-Transferase π
Glutathione S-transferases (GSTs) are an abundant family of detoxification enzymes that catalyze the conjugation of glutathione to chemically reactive electrophilic compounds[39]. The Pi class of GSTs (GSTπ) specifically participates in resistance to drugs and carcinogens. High levels of GSTπ are shown to occur in solid tumors, although their function in the catalytic detoxification process remains ambiguous[40]. A link between GSTπ and S-glutathionylation was first described through the observation that GSTπ could conjugate GSH to peroxiredoxin VI, a non-selenium-dependent lipid peroxidase that converts lipid hydroperoxides to their corresponding alcohols[41].
A critical step in the detoxification reaction catalyzed by peroxiredoxin VI is the formation of the enzyme sulfenic acid on Cys47, the single conserved cysteine residue of peroxiredoxin VI[41]. Cys47 is sterically inaccessible within the enzyme, making the sulfenic acid relatively stable within the globular dimer complex[40]. In addition, the oxidized monomer of the enzyme forms a homodimer that further limits its accessibility[41]. Manevich et al. demonstrated that the heterodimerization of peroxiredoxin VI with GSH-saturated GSTπ resulted in the S-glutathionylation of the oxidized cysteine followed by the subsequent reduction of the mixed disulfide and the activation of the enzyme[41]. These results suggest that GSTπ can facilitate the transfer of GSH to the active site of peroxiredoxin VI through a physiological mechanism that restores the enzymatic activity.
GSTπ has been described to play a key role in the S-glutathionylation mechanism of several proteins, especially during oxidative stress conditions in vivo and in vitro[42][43]. Townsend et al. have shown that GSTπ knockout mice have decreased S-glutathionylated protein levels following PABA/NO treatment and that the rate of S-glutathionylation is significantly enhanced in the presence of GSTπ [40]. In agreement with these findings, in vitro studies using GSTπ-knockdown HEK293 cells have shown decreased levels of S-glutathionylation[44]. Moreover, there is evidence that GSTπ present in the endoplasmic reticulum catalyzes S-glutathionylation of critical proteins within the organelle, providing a potential linkage between redox-based signaling and pathways that regulate the unfolded protein response[45].
Recent studies have identified additional proteins that may also undergo selective S-glutathionylation by GSTπ, including Nrf2 protein inhibitor Keap1 [46], estrogen receptor α[47], actin[48], and AMP-activated protein kinase[49]. Furthermore, it is important to note that GSTπ is also subject to redox regulation[50]. Under oxidative stress, S-glutathionylation on Cys47 and Cys101 is shown to autoregulate GSTπ by oligomerization and enzyme inactivation[40]. Therefore, GSTπ may promote the S-glutathionylation of a wide range of proteins, and its high expression in tumor cells may be related to the aberrant oxidative stress state[51].
3.2. Other Potential Enzymes
Under normal physiological conditions with high levels of GSH, glutaredoxin (Grx) primarily functions in catalyzing the deglutathionylation reaction (i.e., removing glutathione from S-glutathionylated proteins), which is discussed in more detail below. However, during increased levels of oxidative stress, when glutathione is present mainly in its oxidized forms including GSSG, GSNO, and GS•, Grx1 has been proposed to catalyze the forward S-glutathionylation reaction[52]. Mieyal et al. anticipated that due to the low pKa of the active site Cys22 of Grx1 and the stability of disulfide-anion radicals (i.e., Grx1- SSG•−) as opposed to the glutathionyl radical (GS•), Grx1 may catalyze the S-glutathionylation of proteins through a radical intermediate[3]. Grx1 has been shown to promote S-glutathionylation of GAPDH, actin, and PTP1B using glutathionyl radical as the proximal donor[20]. Besides, S-glutathionylation was competitively inhibited by the reaction of O2 with Grx1- SSG•−, confirming that the redox environment likely determines the mode of catalysis by Grx1[20].Recently, a possible role for the glyoxalase II enzyme in the S-glutathionylation of specific proteins has been reported[53][54]. The glyoxalase system, composed of glyoxalases I and II, is involved in the detoxification of methylglyoxal and other reactive aldehydes produced during metabolism[55]. Reduced glutathione is used to convert methylglyoxal to S-D-lactoylglutathione, a thioester of glutathione[55]. The product is then hydrolyzed by glyoxalase II to yield D-lactate and reduced glutathione[55]. Ercolani et al. showed that when the glyoxalase II and S-D-lactoylglutathione were incubated with malate dehydrogenase or with actin, the proteins were S-glutathionylated[53]. At present, a role for glyoxalase II for S-glutathionylation in vivo has not been reported and further investigation is needed to elucidate the potential interaction of the catalytic site of the enzyme with target proteins.
4. Deglutathionylation
Protein S-glutathionylation is a dynamic and reversible process, dependent on both the rate of formation and the rate of deglutathionylation. The reversibility of protein S-glutathionylation provides an additional layer of regulation for controlling cellular processes[56]. Deglutathionylation reactions in cells may occur spontaneously due to the reductive environment and high concentrations of GSH in cells. However, glutaredoxin has specific binding grooves for glutathione and is considered the prime catalyst for deglutathionylation[57]. Glutaredoxin is a member of the thioredoxin superfamily of enzymes, along with thioredoxins, protein disulfide isomerase, glutathione peroxidase, and glutathione S-transferase[58]. Although these proteins show low similarity in their sequence homology, they all incorporate the thioredoxin fold structural motif, which is essential for their redox function[58].
Isoforms of glutaredoxin are found in prokaryotes and the various cellular compartments of eukaryotes. Based on their active site sequences, mammalian glutaredoxins can be broadly categorized into two major subfamilies: class I and class II glutaredoxins[59]. Class I glutaredoxins are localized in the cytosol and mitochondrial intermembrane space, whereas class II glutaredoxins are primarily localized in mitochondria. Class I glutaredoxins are better characterized and have been reported to catalyze most of the deglutathionylating activity in mammalian cells[60][61]. Class II glutaredoxins bind Fe-S clusters and generally exhibit lower oxidoreductase activity than that of class I glutaredoxins[62][63].
The active site of the vast majority of glutaredoxin isoforms contain variations of the redox-active Cys-X-X-Cys motif[64]. The main activity of glutaredoxin involves thiol–disulfide exchange and nucleophilic displacement reactions. A monothiol mechanism and a dithiol mechanism have been proposed, but the monothiol mechanism is generally considered to be the prevalent deglutathionylation mechanism. In the monothiol mechanism, the S-glutathionylated protein thiol is attacked by the thiolate anion of glutaredoxin, forming an enzyme intermediate (Grx-SSG) and releasing the reduced protein. The second step in this mechanism involves the reduction of the enzyme intermediate by GSH to produce GSSG, which is subsequently reduced by glutathione reductase and NADPH. In contrast, the dithiol mechanism uses both active site cysteine residues of glutaredoxin to reduce glutathionylated substrates. Figure 2 depicts a summary of the catalytic mechanism of deglutathionylation by glutaredoxin.
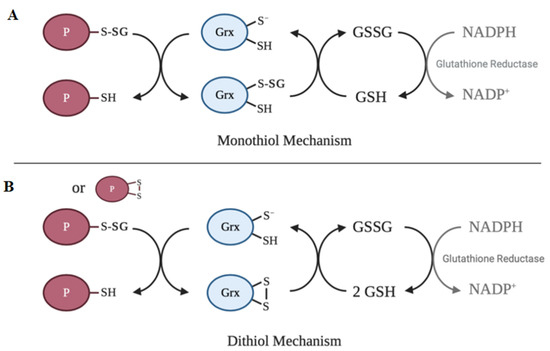
Figure 2. Catalytic mechanisms of deglutathionylation by glutaredoxin. (A) In the monothiol mechanism of deglutathionylation, the thiolate anion of glutaredoxin attacks the S-glutathionylated protein thiol, forming an enzyme-intermediate and releasing the reduced protein. The second step in this mechanism involves the reduction of the enzyme intermediate by GSH to produce GSSG, which is subsequently reduced by glutathione reductase and NADPH. (B) The dithiol mechanism uses both active site cysteine residues of glutaredoxin to reduce glutathionylated substrates. Oxidized glutaredoxin is reduced in the presence of 2 GSH molecules to regenerate the active enzyme.
Another enzyme that has been proposed to have deglutathionylating activity is sulfiredoxin (Srx), a small oxidoreductase that catalyzes the reduction of sulfinic acid derivatives of 2-Cys peroxiredoxins [65]. Findlay et al. observed that the overexpression of Srx in HEK293 cells lowered the levels of S-glutathionylated proteins, such as actin and PTP1B, induced by the diazeniumdiolate PABA/NO [66][67]. Furthermore, Park et al. showed that Srx is specific for deglutathionylating Prx I (known to have a high affinity to Srx) and that the level of S-glutathionylated Prx I was substantially elevated in Srx-knockdown cells [65]. The S-glutathionylation of Prx V, not known to bind to Srx, was not altered by Srx expression levels, demonstrating the specificity of Srx towards Prx I [65]. Although these studies demonstrate that Srx may contribute to the catalysis of deglutathionylation, whether the contribution is significant in vivo is unclear.
References
- Atef Mannaa; Franz-Georg Hanisch; Redox Proteomes in Human Physiology and Disease Mechanisms. Journal of Proteome Research 2019, 19, 1-17, 10.1021/acs.jproteome.9b00586.
- D M Ziegler; Role of Reversible Oxidation-Reduction of Enzyme Thiols-Disulfides in Metabolic Regulation. Annual Review of Biochemistry 1985, 54, 305-329, 10.1146/annurev.bi.54.070185.001513.
- John J. Mieyal; Molly M. Gallogly; Suparna Qanungo; Elizabeth A. Sabens; Melissa D. Shelton; Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxidants & Redox Signaling 2008, 10, 1941-1988, 10.1089/ars.2008.2089.
- Isabella Dalle-Donne; Aldo Milzani; Nicoletta Gagliano; Roberto Colombo; Daniela Giustarini; Ranieri Rossi; Molecular Mechanisms and Potential Clinical Significance ofS-Glutathionylation. Antioxidants & Redox Signaling 2008, 10, 445-474, 10.1089/ars.2007.1716.
- Y.C. Chai; S.S. Ashraf; K. Rokutan; R.B. Johnston; J.A. Thomas; S-Thiolation of Individual Human Neutrophil Proteins Including Actin by Stimulation of the Respiratory Burst: Evidence against a Role for Glutathione Disulfide. Archives of Biochemistry and Biophysics 1994, 310, 273-281, 10.1006/abbi.1994.1167.
- Peter Klatt; Estela Pineda Molina; Mario Garcia Lacoba; C. Alicia Padilla; Emilia Martínez‐Galisteo; J A Barcena; Santiago Lamas; Redox regulation of c‐Jun DNA binding by reversible S‐glutathiolation. The FASEB Journal 1999, 13, 1481-1490, 10.1096/fasebj.13.12.1481.
- Vinayak Gupta; Kate S. Carroll; Sulfenic acid chemistry, detection and cellular lifetime. Biochimica et Biophysica Acta (BBA) - General Subjects 2014, 1840, 847-875, 10.1016/j.bbagen.2013.05.040.
- Mirko Zaffagnini; Mariette Bedhomme; Christophe H. Marchand; Samuel Morisse; Paolo Trost; Stéphane D. Lemaire; Redox Regulation in Photosynthetic Organisms: Focus on Glutathionylation. Antioxidants & Redox Signaling 2012, 16, 567-586, 10.1089/ars.2011.4255.
- Molly M Gallogly; John J Mieyal; Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Current Opinion in Pharmacology 2007, 7, 381-391, 10.1016/j.coph.2007.06.003.
- Jie Wang; Carolyn S Sevier; Formation and Reversibility of BiP Protein Cysteine Oxidation Facilitate Cell Survival during and post Oxidative Stress. Journal of Biological Chemistry 2016, 291, 7541-7557, 10.1074/jbc.m115.694810.
- Seung-Rock Lee; Ki-Sun Kwon; Seung-Ryul Kim; Sue Goo Rhee; Reversible Inactivation of Protein-tyrosine Phosphatase 1B in A431 Cells Stimulated with Epidermal Growth Factor. Journal of Biological Chemistry 1998, 273, 15366-15372, 10.1074/jbc.273.25.15366.
- John M. Denu; Kirk G. Tanner; Specific and Reversible Inactivation of Protein Tyrosine Phosphatases by Hydrogen Peroxide: Evidence for a Sulfenic Acid Intermediate and Implications for Redox Regulation†. Biochemistry 1998, 37, 5633-5642, 10.1021/bi973035t.
- William C. Barrett; Jon P. DeGnore; Simone König; Henry M. Fales; Yen-Fang Keng; Zhong-Yin Zhang; Moon B. Yim; P. Boon Chock; Regulation of PTP1B via Glutathionylation of the Active Site Cysteine 215. Biochemistry 1999, 38, 6699-6705, 10.1021/bi990240v.
- David E. Heppner; Milena Hristova; Christopher M. Dustin; Karamatullah Danyal; Aida Habibovic; Albert Van Der Vliet; The NADPH Oxidases DUOX1 and NOX2 Play Distinct Roles in Redox Regulation of Epidermal Growth Factor Receptor Signaling. Journal of Biological Chemistry 2016, 291, 23282-23293, 10.1074/jbc.m116.749028.
- Mirko Zaffagnini; Christophe H. Marchand; Marco Malferrari; Samuel Murail; Sara Bonacchi; Damiano Genovese; Marco Montalti; Giovanni Venturoli; Giuseppe Falini; Marc Baaden; et al.Stéphane D. LemaireSimona FermaniPaolo Trost Glutathionylation primes soluble glyceraldehyde-3-phosphate dehydrogenase for late collapse into insoluble aggregates. Proceedings of the National Academy of Sciences 2019, 116, 26057-26065, 10.1073/pnas.1914484116.
- Annette Salmeen; Jannik N. Andersen; Michael P. Myers; Tzu-Ching Meng; John A. Hinks; Nicholas K. Tonks; David Barford; Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769-773, 10.1038/nature01680.
- Detcho A. Stoyanovsky; Akihiro Maeda; James L. Atkins; Valerian E. Kagan; Assessments of Thiyl Radicals in Biosystems: Difficulties and New Applications. Analytical Chemistry 2011, 83, 6432-6438, 10.1021/ac200418s.
- Christian Schöneich; Thiyl radicals and induction of protein degradation. Free Radical Research 2015, 50, 143-149, 10.3109/10715762.2015.1077385.
- Madia Trujillo; Beatriz Alvarez; Rafael Radi; One- and two-electron oxidation of thiols: mechanisms, kinetics and biological fates. Free Radical Research 2015, 50, 150-171, 10.3109/10715762.2015.1089988.
- David W. Starke; P. Boon Chock; John J. Mieyal; Glutathione-Thiyl Radical Scavenging and Transferase Properties of Human Glutaredoxin (Thioltransferase). Journal of Biological Chemistry 2003, 278, 14607-14613, 10.1074/jbc.m210434200.
- Patrick T. Kang; Liwen Zhang; Chwen-Lih Chen; Jingfeng Chen; Kari B. Green-Church; Yeong-Renn Chen; Protein thiyl radical mediates S-glutathionylation of complex I. Free Radical Biology and Medicine 2012, 53, 962-973, 10.1016/j.freeradbiomed.2012.05.025.
- Patrick T. Kang; Chwen-Lih Chen; Yeong-Renn Chen; Increased mitochondrial prooxidant activity mediates up-regulation of Complex I S-glutathionylation via protein thiyl radical in the murine heart of eNOS−/−. Free Radical Biology and Medicine 2015, 79, 56-68, 10.1016/j.freeradbiomed.2014.11.016.
- Chun-An Chen; Cho-Hao Lin; Lawrence J. Druhan; Tse-Yao Wang; Yeong-Renn Chen; Jay L. Zweier; Superoxide Induces Endothelial Nitric-oxide Synthase Protein Thiyl Radical Formation, a Novel Mechanism Regulating eNOS Function and Coupling. Journal of Biological Chemistry 2011, 286, 29098-29107, 10.1074/jbc.m111.240127.
- Jay L. Zweier; Chun-An Chen; Lawrence J. Druhan; S-Glutathionylation Reshapes Our Understanding of Endothelial Nitric Oxide Synthase Uncoupling and Nitric Oxide/Reactive Oxygen Species-Mediated Signaling. Antioxidants & Redox Signaling 2011, 14, 1769-1775, 10.1089/ars.2011.3904.
- Matthew W. Foster; Douglas T. Hess; Jonathan S. Stamler; Protein S-nitrosylation in health and disease: a current perspective. Trends in Molecular Medicine 2009, 15, 391-404, 10.1016/j.molmed.2009.06.007.
- Pamela Ehrenfeld; Francisco Cordova; Walter N. Duran; Fabiola A. Sánchez; S-nitrosylation and its role in breast cancer angiogenesis and metastasis. Nitric Oxide 2019, 87, 52-59, 10.1016/j.niox.2019.03.002.
- Colin T. Stomberski; Douglas T. Hess; Jonathan S. Stamler; Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxidants & Redox Signaling 2019, 30, 1331-1351, 10.1089/ars.2017.7403.
- D. Lyn H. Williams; The Chemistry of S-Nitrosothiols. Accounts of Chemical Research 1999, 32, 869-876, 10.1021/ar9800439.
- Christina L. Grek; Jie Zhang; Yefim Manevich; Danyelle M. Townsend; Kenneth D. Tew; Causes and Consequences of CysteineS-Glutathionylation. Journal of Biological Chemistry 2013, 288, 26497-26504, 10.1074/jbc.r113.461368.
- Eugenia Belcastro; Caroline Gaucher; Alessandro Corti; Pierre Leroy; Isabelle Lartaud; Alfonso Pompella; Regulation of protein function by S-nitrosation and S-glutathionylation: processes and targets in cardiovascular pathophysiology. Biological Chemistry 2017, 398, 1267-1293, 10.1515/hsz-2017-0150.
- Peter Klatt; Estela Pineda-Molina; Santiago Lamas; Nitric Oxide Inhibits c-Jun DNA Binding by Specifically TargetedS-Glutathionylation. Journal of Biological Chemistry 1999, 274, 15857-15864, 10.1074/jbc.274.22.15857.
- Daniela Giustarini; Aldo Milzani; Giancarlo Aldini; Marina Carini; Ranieri Rossi; Isabella Dalle-Donne; S-Nitrosation versus S-Glutathionylation of Protein Sulfhydryl Groups by S-Nitrosoglutathione. Antioxidants & Redox Signaling 2005, 7, 930-939, 10.1089/ars.2005.7.930.
- Susanne Mohr; Hazem Hallak; Alexander De Boitte; Eduardo G. Lapetina; Bernhard Brüne; Nitric Oxide-inducedS-Glutathionylation and Inactivation of Glyceraldehyde-3-phosphate Dehydrogenase. Journal of Biological Chemistry 1999, 274, 9427-9430, 10.1074/jbc.274.14.9427.
- T. L. Dutka; J. P. Mollica; C. R. Lamboley; V. C. Weerakkody; D. W. Greening; G. S. Posterino; Robyn M. Murphy; Graham D. Lamb; S-nitrosylation and S-glutathionylation of Cys134 on troponin I have opposing competitive actions on Ca2+ sensitivity in rat fast-twitch muscle fibers. American Journal of Physiology-Cell Physiology 2017, 312, C316-C327, 10.1152/ajpcell.00334.2016.
- Kuo-Ping Huang; Freesia L Huang; Glutathionylation of proteins by glutathione disulfide S-oxide. Biochemical Pharmacology 2002, 64, 1049-1056, 10.1016/s0006-2952(02)01175-9.
- Junfa Li; Freesia L. Huang; Kuo-Ping Huang; Glutathiolation of Proteins by Glutathione DisulfideS-Oxide Derived fromS-Nitrosoglutathione. Journal of Biological Chemistry 2000, 276, 3098-3105, 10.1074/jbc.m008260200.
- Tatsuya Okamoto; Takaaki Akaike; Tomohiro Sawa; Yoichi Miyamoto; Albert Van Der Vliet; Hiroshi Maeda; Activation of Matrix Metalloproteinases by Peroxynitrite-induced ProteinS-Glutathiolation via DisulfideS-Oxide Formation. Journal of Biological Chemistry 2001, 276, 29596-29602, 10.1074/jbc.m102417200.
- Mahdieh Sadidi; Timothy J. Geddes; Donald M. Kuhn; S-Thiolation of Tyrosine Hydroxylase by Reactive Nitrogen Species in the Presence of Cysteine or Glutathione. Antioxidants & Redox Signaling 2005, 7, 863-869, 10.1089/ars.2005.7.863.
- E. Boyland; L. F. Chasseaud; Enzyme-catalysed conjugations of glutathione with unsaturated compounds. Biochemical Journal 1967, 104, 95-102, 10.1042/bj1040095.
- Danyelle M. Townsend; Yefim Manevich; Lin He; Steven Hutchens; Christopher J. Pazoles; Kenneth D. Tew; Novel Role for GlutathioneS-Transferase π. Journal of Biological Chemistry 2008, 284, 436-445, 10.1074/jbc.m805586200.
- Y. Manevich; S. I. Feinstein; Aron B. Fisher; Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with GST. Proceedings of the National Academy of Sciences 2004, 101, 3780-3785, 10.1073/pnas.0400181101.
- D. Bartolini; P. Torquato; M. Piroddi; F. Galli; Targeting glutathione S-transferase P and its interactome with selenium compounds in cancer therapy. Biochimica et Biophysica Acta (BBA) - General Subjects 2018, 1863, 130-143, 10.1016/j.bbagen.2018.09.023.
- Danyelle M. Townsend; Lin He; Steven Hutchens; Tracy E. Garrett; Christopher J. Pazoles; Kenneth D. Tew; NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance.. Cancer Research 2008, 68, 2870-7, 10.1158/0008-5472.CAN-07-5957.
- Kwan Young Ko; Jea Hwang Lee; Jun Ki Jang; Yunjung Jin; Hyunwoo Kang; Ick Young Kim; S-Glutathionylation of mouse selenoprotein W prevents oxidative stress-induced cell death by blocking the formation of an intramolecular disulfide bond. Free Radical Biology and Medicine 2019, 141, 362-371, 10.1016/j.freeradbiomed.2019.07.007.
- Zhi-Wei Ye; Jie Zhang; Tiffany Ancrum; Yefim Manevich; Danyelle M. Townsend; Kenneth D. Tew; Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxidants & Redox Signaling 2017, 26, 247-261, 10.1089/ars.2015.6486.
- Andreia N Carvalho; Carla Marques; Rita C Guedes; Margarida Castro-Caldas; Elsa Rodrigues; Jack Van Horssen; M. J. Gama; S-Glutathionylation of Keap1: a new role for glutathioneS-transferase pi in neuronal protection. FEBS Letters 2016, 590, 1455-1466, 10.1002/1873-3468.12177.
- Jie Zhang; Zhi-Wei Ye; Wei Chen; Y. Manevich; Shikhar Mehrotra; Lauren E. Ball; Yvonne M Janssen-Heininger; Kenneth D. Tew; Danyelle M. Townsend; S-Glutathionylation of estrogen receptor α affects dendritic cell function. Journal of Biological Chemistry 2018, 293, 4366-4380, 10.1074/jbc.m117.814327.
- Takeshi Uemura; George Tsaprailis; Eugene W. Gerner; Hygor Ferreira-Fernandes; France K. Yoshioka; Alcemir R. Santos; Helem F. Ribeiro; Juan A. Rey; Paulo C. Soares; Rommel R. Burbano; et al.Giovanny R. Pinto GSTΠ stimulates caveolin-1-regulated polyamine uptake via actin remodeling. Oncotarget 2019, 10, 5713-5723, 10.18632/oncotarget.27192.
- Anna Klaus; Sarah Zorman; Alexandre Berthier; Cécile Polge; Sacnicte Ramirez; Sylvie Michelland; Michel Sève; Didier Vertommen; Mark Rider; Nicolas Lentze; et al.Daniel AuerbachUwe Schlattner Glutathione S-Transferases Interact with AMP-Activated Protein Kinase: Evidence for S-Glutathionylation and Activation In Vitro. PLOS ONE 2013, 8, e62497, 10.1371/journal.pone.0062497.
- Kenneth D. Tew; Danyelle M. Townsend; Regulatory functions of glutathioneS-transferase P1-1 unrelated to detoxification. Drug Metabolism Reviews 2011, 43, 179-193, 10.3109/03602532.2011.552912.
- Naoki Fujitani; Akihiro Yoneda; Motoko Takahashi; Akira Takasawa; Tomoyuki Aoyama; Tadaaki Miyazaki; Silencing of Glutathione S-Transferase Pi Inhibits Cancer Cell Growth via Oxidative Stress Induced by Mitochondria Dysfunction.. Scientific Reports 2019, 9, 14764-13, 10.1038/s41598-019-51462-9.
- Yvonne M. W. Janssen-Heininger; James D. Nolin; Sidra M. Hoffman; Jos L. Van Der Velden; Jane E. Tully; Karolyn G. Lahue; Sarah T. Abdalla; David G. Chapman; Niki L. Reynaert; Albert Van Der Vliet; et al.Vikas Anathy Emerging mechanisms of glutathione-dependent chemistry in biology and disease. Journal of Cellular Biochemistry 2013, 114, 1962-1968, 10.1002/jcb.24551.
- Luisa Ercolani; Andrea Scirè; Roberta Galeazzi; Luca Massaccesi; Laura Cianfruglia; Adolfo Amici; Francesco Piva; Lorena Urbanelli; Carla Emiliani; Giovanni Principato; et al.Tatiana Armeni A possible S-glutathionylation of specific proteins by glyoxalase II: An in vitro and in silico study. Cell Biochemistry and Function 2016, 34, 620-627, 10.1002/cbf.3236.
- Roberta Galeazzi; Emiliano Laudadio; Emanuele Falconi; Luca Massaccesi; Luisa Ercolani; Giovanna Mobbili; Cristina Minnelli; Andrea Scirè; Laura Cianfruglia; Tatiana Armeni; et al. Protein–protein interactions of human glyoxalase II: findings of a reliable docking protocol. Organic & Biomolecular Chemistry 2017, 16, 5167-5177, 10.1039/c8ob01194j.
- Sebastian Brings; Thomas Fleming; Marc Freichel; M U Muckenthaler; Stephan Herzig; Peter Nawroth; Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. International Journal of Molecular Sciences 2017, 18, 984, 10.3390/ijms18050984.
- Robert Harmel; Dorothea Fiedler; Features and regulation of non-enzymatic post-translational modifications. Nature Chemical Biology 2018, 14, 244-252, 10.1038/nchembio.2575.
- Reiko Matsui; Beatriz Ferran; Albin Oh; Dominique Croteau; Di Shao; Jingyan Han; David Richard Pimentel; Markus Michael Bachschmid; Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxidants & Redox Signaling 2020, 32, 677-700, 10.1089/ars.2019.7963.
- Guoping Ren; Daniel Stephan; Zhaohui Xu; Ying Zheng; Danming Tang; Rosemary S. Harrison; Mareike Kurz; Russell J. Jarrott; Stephen R. Shouldice; Annie Hiniker; et al.Jennifer L. MartinBegoña HerasJames C. A. Bardwell Properties of the Thioredoxin Fold Superfamily Are Modulated by a Single Amino Acid Residue. Journal of Biological Chemistry 2009, 284, 10150-10159, 10.1074/jbc.m809509200.
- Jannik Zimmermann; Julian Oestreicher; Steffen Hess; Johannes M. Herrmann; Marcel DePonte; Bruce Morgan; One cysteine is enough: A monothiol Grx can functionally replace all cytosolic Trx and dithiol Grx. Redox Biology 2020, 36, 101598, 10.1016/j.redox.2020.101598.
- Carol A. Chrestensen; David W. Starke; John J. Mieyal; Acute Cadmium Exposure Inactivates Thioltransferase (Glutaredoxin), Inhibits Intracellular Reduction of Protein-glutathionyl-mixed Disulfides, and Initiates Apoptosis. Journal of Biological Chemistry 2000, 275, 26556-26565, 10.1074/jbc.m004097200.
- Che-Hun Jung; James A. Thomas; S-Glutathiolated Hepatocyte Proteins and Insulin Disulfides as Substrates for Reduction by Glutaredoxin, Thioredoxin, Protein Disulfide Isomerase, and Glutathione. Archives of Biochemistry and Biophysics 1996, 335, 61-72, 10.1006/abbi.1996.0482.
- E. Herrero; M. A. De La Torre-Ruiz; Monothiol glutaredoxins: a common domain for multiple functions. Cellular and Molecular Life Sciences 2007, 64, 1518-1530, 10.1007/s00018-007-6554-8.
- Jérémy Couturier; Jonathan Przybyla-Toscano; Thomas Roret; Claude Didierjean; Nicolas Rouhier; The roles of glutaredoxins ligating Fe–S clusters: Sensing, transfer or repair functions?. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2015, 1853, 1513-1527, 10.1016/j.bbamcr.2014.09.018.
- Carsten Berndt; Christopher Horst Lillig; Arne Holmgren; Thioredoxins and glutaredoxins as facilitators of protein folding. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2008, 1783, 641-650, 10.1016/j.bbamcr.2008.02.003.
- Ji Won Park; John J. Mieyal; Sue Goo Rhee; P. Boon Chock; Deglutathionylation of 2-Cys Peroxiredoxin Is Specifically Catalyzed by Sulfiredoxin. Journal of Biological Chemistry 2009, 284, 23364-23374, 10.1074/jbc.m109.021394.
- Victoria J. Findlay; Haim Tapiero; Danyelle M. Townsend; Sulfiredoxin: a potential therapeutic agent?. Biomedicine & Pharmacotherapy 2005, 59, 374-379, 10.1016/j.biopha.2005.07.003.
- Victoria J. Findlay; Danyelle M. Townsend; Taylor E. Morris; Jacob P. Fraser; Lin He; Kenneth D. Tew; A novel role for human sulfiredoxin in the reversal of glutathionylation.. Cancer Research 2006, 66, 6800-6, 10.1158/0008-5472.CAN-06-0484.

