 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dean Liu | -- | 1240 | 2022-10-14 01:32:46 |
Video Upload Options
The Henry Reaction (also referred to as the nitro-aldol reaction) is a classic carbon–carbon bond formation reaction in organic chemistry. Discovered in 1895 by the Belgian chemist Louis Henry (1834-1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-Nitro alcohols. This type of reaction is commonly referred to as a "nitro-aldol" reaction (nitroalkane, aldehyde, and alcohol) It is nearly analogous to the aldol reaction that had been discovered 23 years prior that couples two carbonyl compounds to form β-hydroxy carbonyl compounds known as "aldols" (aldehyde and alcohol). The Henry reaction is a useful technique in the area of organic chemistry due to the synthetic utility of its corresponding products, as they can be easily converted to other useful synthetic intermediates. These conversions include subsequent dehydration to yield nitroalkenes, oxidation of the secondary alcohol to yield α-nitro ketones, or reduction of the nitro group to yield β-amino alcohols. Many of these uses have been exemplified in the syntheses of various pharmaceuticals including the β-blocker (S)-propranolol, the HIV protease inhibitor Amprenavir (Vertex 478), and construction of the carbohydrate subunit of the anthracycline class of antibiotics, L-Acosamine. The synthetic scheme of the L-Acosamine synthesis can be found in the Examples section of this article.
1. Mechanism
The Henry reaction begins with the deprotonation of the nitroalkane on the α-carbon position forming a resonance stabilized anion. The pKa of most nitroalkanes is approximately 17.[1][2] This is followed by alkylation of the nitroalkane with the carbonyl containing substrate to form a diastereomeric β-nitro alkoxide. The protonation of the alkoxide by the previously protonated base will yield the respective β-nitro alcohol as product. (Scheme below)
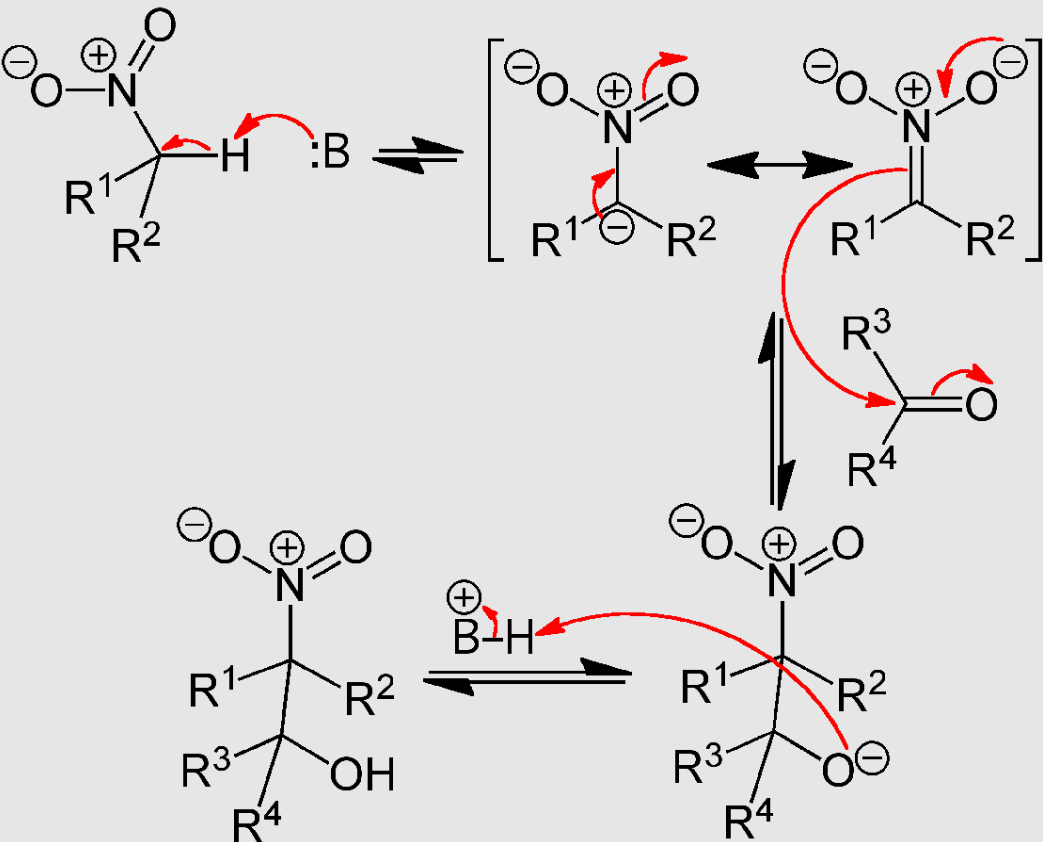
It is important to note that all steps of the Henry reaction are reversible. This is due to the lack of a committed step in the reaction to form product. It is for this reason that research has been geared towards modifications to drive the reaction to completion.[3][4] More information about this can be found in the modification section of this article.
2. Stereochemical Course
One of the commonly accepted models for stereoselection without any modification to the Henry reaction is shown below where stereoselectivity is governed by the size of the R groups in the model (ex. carbon chain) as well as a transition state that minimizes dipole by orienting the nitro group and carbonyl oxygen anti (on opposite sides) each other. The R groups play a role in the transition state of the Henry reaction in that the larger the R groups are on each of the substrates, the more they will want to orient themselves away from each other (commonly referred to as steric effects) [4][5]
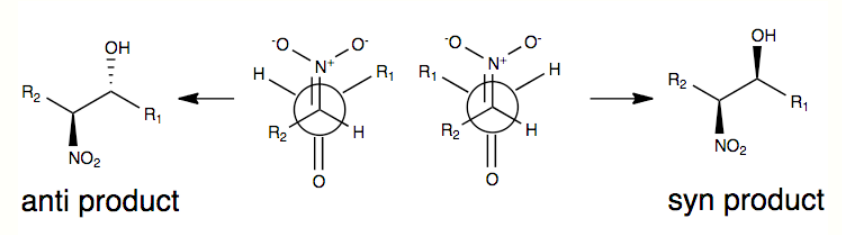
Due to a number of factors, including the reversibility of the reaction, as well as the tendency for easy epimerization of the nitro-substituted carbon atom, the Henry Reaction will typically produce a mixture of enantiomers or diastereomers. It is for this reason that explanations for stereoselectivity remain scarce without some modification.[4] In recent years, research focus has shifted toward modifications of the Henry Reaction to overcome this synthetic challenge.
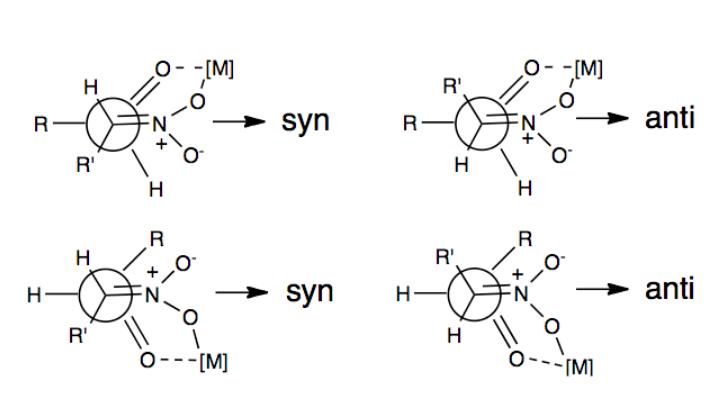
The first example of an enatioselective nitroaldol reaction was reported in 1992 using Shibasaki catalysts.[6] One of the most frequently employed ways to induce enantio- or diastereoselectivity in the Henry Reaction has been through the use of chiral metal catalysts in which the nitro group and carbonyl oxygen coordinate to a metal that is bound to a chiral organic molecule. Some examples of metals that have been used include Zn, Co, Cu, Mg, and Cr.[7] A depiction of this coordination is illustrated above.
3. General Features
One of the many features of the Henry Reaction that makes it synthetically attractive is that it utilizes only a catalytic amount of base to drive the reaction. Additionally a variety of bases can be used including ionic bases such as alkali metal hydroxides, alkoxides, carbonates, and sources of fluoride anion (e.g. TBAF) or nonionic organic amine bases including TMG, DBU, DBN, and PAP. It is important to note that the base and solvent used do not have a large influence on the overall outcome of the reaction.[3]
4. Limitations
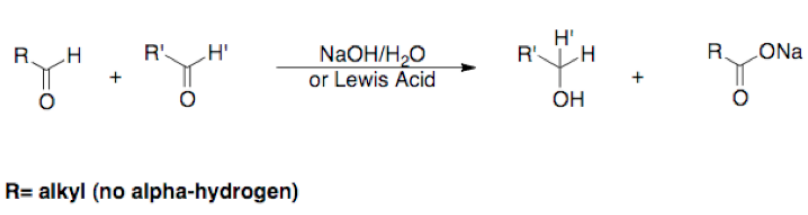
One of the main drawbacks of the Henry Reaction is the potential for side reactions throughout the course of the reaction. Aside from the reversibility of the reaction (Retro-Henry) which could prevent the reaction from proceeding, the β-nitro alcohol has the potential to undergo dehydration, and for sterically hindered substrates it is possible that a base catalyzed self-condensation (Cannizaro reaction) could occur. A general scheme of the Cannizzaro reaction is depicted below.[3]
5. Modifications
There have been a series of modifications made to the Henry Reaction. Of these some of the most important include employing high-pressure and sometimes solvent free conditions to improve chemo- and regioselectivity[3] and chiral metal catalysts to induce enantio-or diastereoselectivity.[7] The aza-Henry reaction is also used to produce nitroamines and can be a reliable synthetic route for the synthesis of vicinal diamines.[8]
Perhaps one of the most synthetically useful modifications to the Henry Reaction is the use of an organocatalyst.[3][7][9] The catalytic cycle is shown below.

List described that while this is a broad explanation, his brief review illustrates that this is a plausible mechanistic explanation for almost all reactions that involve an organocatalyst. An example of this type of reaction is illustrated in the Examples section of this article.
In addition to the previously mentioned modifications to the Henry reaction there are a variety of others. This includes the conversion of unreactive alkyl nitro compounds to their corresponding dianions which will react faster with carbonyl substrates, reactions can be accelerated using PAP as base, utilization of the reactivity of aldehydes with α,α-doubly deprotonated nitroalkanes to give nitronate alkoxides that yield mainly syn-nitro alcohols once protonated, and finally generation of nitronate anions in which one oxygenatom on the nitro group is silyl-protected to yield anti-β-nitro alcohols in the presence of a fluoride anion source when reacted with an aldehyde.[3][4]
6. Examples
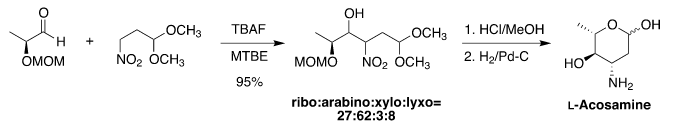
Industrial Application- In 1999, Menzel and coworkers developed a synthetic route to obtaining L-Acosamine, the carbohydrate subunit of the anthracycline class of antibiotics[10][11]
Industrial Application- An enantioselective aldol addition product can be obtained in asymmetric synthesis by reaction of benzaldehyde with nitromethane and the a catalyst system consisting of a zinc triflate salt / the base diisopropylethylamine (DIPEA) and as chiral ligand is the N-methyl derivative of (+)-ephedrine (NME).[12]
A diastereoselective variation of this reaction is depicted below.[13]
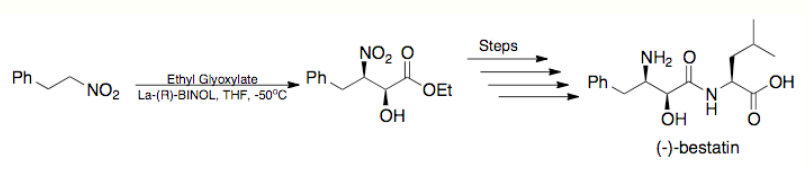
Total Synthesis- In 2005, Barua and coworkers completed the total synthesis of the potent aminopeptidase inhibitor, (–)-bestatin, in an overall yield of 26% overall yield employing Shibasaki's asymmetric Henry reaction as the key step. (illustrated below)[10][14]
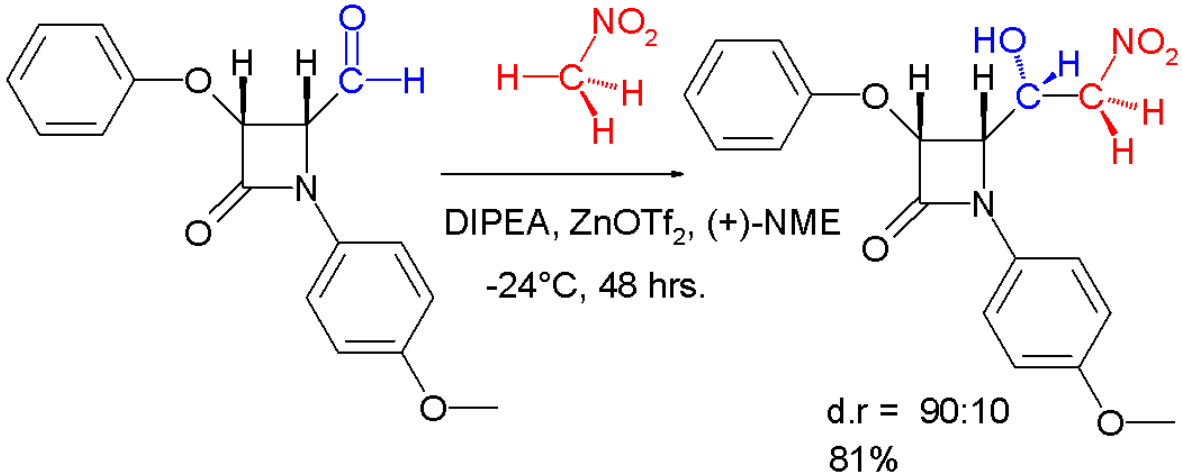
Organocatalysis- In 2006, Hiemstra and coworkers explored the use of quinine derivatives as asymmetric catalysts for the reaction between aromatic aldehydes and nitromethane. Through the use of particular derivatives, they were able to induce direct enantioselection through the use of the proper catalyst.[15]
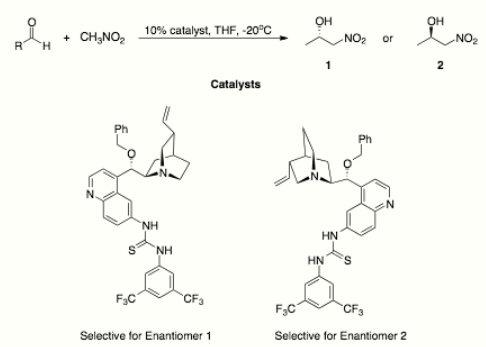
Biocatalysis - In 2006, Purkarthofer et al. found that (S)-hydroxynitrile lyase from Hevea brasiliensis catalyzes the formation of (S)-β-nitro alcohols.[16] In 2011, Fuhshuku and Asano showed that the (R)-selective hydroxynitrile lyase from Arabidopsis thaliana could catalyze the synthesis of (R)-β-nitro alcohols from nitromethane and aromatic aldehydes.[17]
References
- Reich, Hans. "Bordwell pKa table: "Nitroalkanes"". http://www.chem.wisc.edu/areas/reich/pkatable/. Retrieved 17 January 2016.
- Matthews, Walter (1975). "Equilibrium acidities of carbon acids. VI. Establishment of an absolute scale of acidities in dimethyl sulfoxide solution". Journal of the American Chemical Society 97 (24): 7006. doi:10.1021/ja00857a010. http://pubs.acs.org/doi/abs/10.1021/ja00857a010. Retrieved 17 January 2016.
- Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington, MA: Elsevier Academic Press. pp. 202–203. ISBN 0-12-369483-3.
- Noboro, Ono (2001). The Nitro Group in Organic Synthesis. New York, NY: Wiley-VCH. pp. 30–69. ISBN 0-471-31611-3.
- Begona, L., Arrieta, A., Morao, I., Cossio, F.P. (1997). "Ab Initio Models for the Nitroaldol (Henry) Reaction". Chem. Eur. J. 3 (1): 20–28. doi:10.1002/chem.19970030105. https://dx.doi.org/10.1002%2Fchem.19970030105
- Sasai, Hiroaki; Suzuki, Takeyuki; Arai, Shigeru; Arai, Takayoshi; Shibasaki, Masakatsu (1 May 1992). "Basic character of rare earth metal alkoxides. Utilization in catalytic carbon-carbon bond-forming reactions and catalytic asymmetric nitroaldol reactions". Journal of the American Chemical Society 114 (11): 4418–4420. doi:10.1021/ja00037a068. https://dx.doi.org/10.1021%2Fja00037a068
- . List et al. described this process as the organocatalyst functioning as Lewis acid or base or Bronsted Acid or Base.
- Westermann, B. (2003). "Asymmetric catalytic aza-Henry reactions leading to 1,2-diamines and 1,2-diaminocarboxylic acids". Angew. Chem. Int. Ed. Engl. 42: 151–153. doi:10.1002/anie.200390071. https://dx.doi.org/10.1002%2Fanie.200390071
- Seayad, J., List, B. (2005). "Asymmetric organocatalysis". Org. Biomol. Chem. 3 (5): 719–724. doi:10.1039/b415217b. PMID 15731852. https://dx.doi.org/10.1039%2Fb415217b
- Luzzio, F.A. (2001). "The Henry Reaction: recent examples". Tetrahedron 57 (22): 915–945. doi:10.1002/chin.200122233. https://dx.doi.org/10.1002%2Fchin.200122233
- Menzel, A., Ohrlein, R., Griesser, H., Wehner, V., Jager, V. (1999). "A Short Synthesis of L-Acosamine Based on Nitroaldol Addition (Henry Reaction). Analysis of the Key Step Concerning Solvent and Temperature Effects". Synthesis 9 (45): 1691–1702. doi:10.1002/chin.199945325. https://dx.doi.org/10.1002%2Fchin.199945325
- Palomo, Claudio; Oiarbide, Mikel; Laso, Antonio (2005). "Enantioselective Henry Reactions under Dual Lewis Acid/Amine Catalysis Using Chiral Amino Alcohol Ligands". Angewandte Chemie 44 (25): 3881–3884. doi:10.1002/anie.200463075. http://www3.interscience.wiley.com/cgi-bin/abstract/110494368/ABSTRACT.
- Alcaide, Benito; Almendros, Pedro; Luna, Amparo; Paz de Arriba, M.; Rosario Torresc, M. (2007). "Organocatalyzed diastereoselective Henry reaction of enantiopure 4-oxoazetidine-2-carbaldehydes". ARKIVOC 2007 (iv): 285–296. doi:10.3998/ark.5550190.0008.425. http://www.arkat-usa.org/ARKIVOC/JOURNAL_CONTENT/manuscripts/2007/JB-2021JP%20as%20published%20mainmanuscript.pdf.
- Gogoi, N., Boruwa, J., Barua, N.C. (2005). "A total synthesis of (–)-bestatin using Shibasaki's asymmetric Henry reaction". Tetrahedron Letters 46 (44): 7581–7582. doi:10.1016/j.tetlet.2005.08.153. https://dx.doi.org/10.1016%2Fj.tetlet.2005.08.153
- Marcelli, T., van der Haas, R., van Maarseveen, J.H., Hiemstra, H. (2006). "Asymmetric Organocatalytic Henry Reaction". Angew. Chem. Int. Ed. 45 (6): 929–931. doi:10.1002/anie.200503724. https://dx.doi.org/10.1002%2Fanie.200503724
- Purkarthofer, T., Gruber, K., Gruber-Khadjawi, M., Waich, K., Skranc, W., Mink, D. and Griengl, H. (2006). "A Biocatalytic Henry Reaction—The Hydroxynitrile Lyase from Hevea brasiliensis Also Catalyzes Nitroaldol Reactions.". Angewandte Chemie 45: 3454–3456.. doi:10.1002/anie.200504230. https://dx.doi.org/10.1002%2Fanie.200504230
- "Synthesis of (R)-β-nitro alcohols catalyzed by R-selective hydroxynitrile lyase from Arabidopsis thaliana in the aqueous-organic biphasic system.". J. Biotechnol. 153: 153–9.. 2011. doi:10.1016/j.jbiotec.2011.03.011. PMID 21439333. https://dx.doi.org/10.1016%2Fj.jbiotec.2011.03.011


