 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dean Liu | -- | 7126 | 2022-10-14 01:49:16 |
Video Upload Options
Electrochemistry is the branch of physical chemistry concerned with the relationship between electrical potential difference, as a measurable and quantitative phenomenon, and identifiable chemical change, with the potential difference as an outcome of a particular chemical change, or vice versa. These reactions involve electrons moving via an electronically-conducting phase (typically an external electrical circuit, but not necessarily, as in electroless plating) between electrodes separated by an ionically conducting and electronically insulating electrolyte (or ionic species in a solution). When a chemical reaction is driven by an electrical potential difference, as in electrolysis, or if a potential difference results from a chemical reaction as in a battery or fuel cell, it is called an electrochemical reaction. Unlike in other chemical reactions, in electrochemical reactions electrons are not transferred directly between atoms, ions, or molecules, but via the aforementioned electronically-conducting circuit. This phenomenon is what distinguishes an electrochemical reaction from a conventional chemical reaction.
1. History
16th-to-18th-century developments
Understanding of electrical matters began in the sixteenth century. During this century, the English scientist William Gilbert spent 17 years experimenting with magnetism and, to a lesser extent, electricity. For his work on magnets, Gilbert became known as the "Father of Magnetism." He discovered various methods for producing and strengthening magnets.[1]
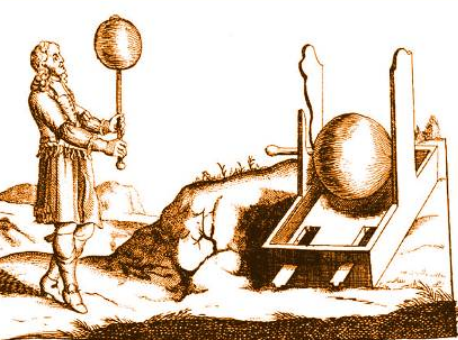
In 1663, the Germany physicist Otto von Guericke created the first electric generator, which produced static electricity by applying friction in the machine. The generator was made of a large sulfur ball cast inside a glass globe, mounted on a shaft. The ball was rotated by means of a crank and an electric spark was produced when a pad was rubbed against the ball as it rotated. The globe could be removed and used as source for experiments with electricity.[2]
By the mid-18th century the France chemist Charles François de Cisternay du Fay had discovered two types of static electricity, and that like charges repel each other whilst unlike charges attract. Du Fay announced that electricity consisted of two fluids: "vitreous" (from the Latin for "glass"), or positive, electricity; and "resinous," or negative, electricity. This was the two-fluid theory of electricity, which was to be opposed by Benjamin Franklin's one-fluid theory later in the century.[3]

In 1785, Charles-Augustin de Coulomb developed the law of electrostatic attraction as an outgrowth of his attempt to investigate the law of electrical repulsions as stated by Joseph Priestley in England.[4]

In the late 18th century the Italian physician and anatomist Luigi Galvani marked the birth of electrochemistry by establishing a bridge between chemical reactions and electricity on his essay "De Viribus Electricitatis in Motu Musculari Commentarius" (Latin for Commentary on the Effect of Electricity on Muscular Motion) in 1791 where he proposed a "nerveo-electrical substance" on biological life forms.[5]
In his essay Galvani concluded that animal tissue contained a here-to-fore neglected innate, vital force, which he termed "animal electricity," which activated nerves and muscles spanned by metal probes. He believed that this new force was a form of electricity in addition to the "natural" form produced by lightning or by the electric eel and torpedo ray as well as the "artificial" form produced by friction (i.e., static electricity).[6]
Galvani's scientific colleagues generally accepted his views, but Alessandro Volta rejected the idea of an "animal electric fluid," replying that the frog's legs responded to differences in metal temper, composition, and bulk.[5][6] Galvani refuted this by obtaining muscular action with two pieces of the same material. Nevertheless, Volta's experimentation led him to develop the first practical battery, which took advantage of the relatively high energy (weak bonding) of zinc and could deliver an electrical current for much longer than any other device known at the time.
19th century

In 1800, William Nicholson and Johann Wilhelm Ritter succeeded in decomposing water into hydrogen and oxygen by electrolysis using Volta's battery. Soon thereafter Ritter discovered the process of electroplating. He also observed that the amount of metal deposited and the amount of oxygen produced during an electrolytic process depended on the distance between the electrodes.[7] By 1801, Ritter observed thermoelectric currents and anticipated the discovery of thermoelectricity by Thomas Johann Seebeck.[8]
By the 1810s, William Hyde Wollaston made improvements to the galvanic cell. Sir Humphry Davy's work with electrolysis led to the conclusion that the production of electricity in simple electrolytic cells resulted from chemical action and that chemical combination occurred between substances of opposite charge. This work led directly to the isolation of metallic sodium and potassium by electrolysis of their molten salts, and of the alkaline earth metals from theirs, in 1808.[9]
Hans Christian Ørsted's discovery of the magnetic effect of electric currents in 1820 was immediately recognized as an epoch-making advance, although he left further work on electromagnetism to others. André-Marie Ampère quickly repeated Ørsted's experiment, and formulated them mathematically.[10]
In 1821, Estonian-German physicist Thomas Johann Seebeck demonstrated the electrical potential between the juncture points of two dissimilar metals when there is a temperature difference between the joints.[11]
In 1827, the German scientist Georg Ohm expressed his law in this famous book "Die galvanische Kette, mathematisch bearbeitet" (The Galvanic Circuit Investigated Mathematically) in which he gave his complete theory of electricity.[11]
In 1832, Michael Faraday's experiments led him to state his two laws of electrochemistry. In 1836, John Daniell invented a primary cell which solved the problem of polarization by introducing copper ions into the solution near the positive electrode and thus eliminating hydrogen gas generation. Later results revealed that at the other electrode, amalgamated zinc (i.e., zinc alloyed with mercury) would produce a higher voltage.
William Grove produced the first fuel cell in 1839. In 1846, Wilhelm Weber developed the electrodynamometer. In 1868, Georges Leclanché patented a new cell which eventually became the forerunner to the world's first widely used battery, the zinc–carbon cell.[7]
Svante Arrhenius published his thesis in 1884 on Recherches sur la conductibilité galvanique des électrolytes (Investigations on the galvanic conductivity of electrolytes). From his results the author concluded that electrolytes, when dissolved in water, become to varying degrees split or dissociated into electrically opposite positive and negative ions.[12]
In 1886, Paul Héroult and Charles M. Hall developed an efficient method (the Hall–Héroult process) to obtain aluminium using electrolysis of molten alumina.[13]
In 1894, Friedrich Ostwald concluded important studies of the conductivity and electrolytic dissociation of organic acids.[14]

Walther Hermann Nernst developed the theory of the electromotive force of the voltaic cell in 1888. In 1889, he showed how the characteristics of the voltage produced could be used to calculate the free energy change in the chemical reaction producing the voltage. He constructed an equation, known as Nernst equation, which related the voltage of a cell to its properties.[15]
In 1898, Fritz Haber showed that definite reduction products can result from electrolytic processes if the potential at the cathode is kept constant. In 1898, he explained the reduction of nitrobenzene in stages at the cathode and this became the model for other similar reduction processes.[16]
20th century and recent developments
In 1902, The Electrochemical Society (ECS) was founded.[17]
In 1909, Robert Andrews Millikan began a series of experiments (see oil drop experiment) to determine the electric charge carried by a single electron.[18] In 1911, Harvey Fletcher, working with Millikan, was successful in measuring the charge on the electron, by replacing the water droplets used by Millikan, which quickly evaporated, with oil droplets. Within one day Fletcher measured the charge of an electron within several decimal places[19]
In 1923, Johannes Nicolaus Brønsted and Martin Lowry published essentially the same theory about how acids and bases behave, using an electrochemical basis.[20]
In 1937, Arne Tiselius developed the first sophisticated electrophoretic apparatus. Some years later, he was awarded the 1948 Nobel Prize for his work in protein electrophoresis.[21]
A year later, in 1949, the International Society of Electrochemistry (ISE) was founded.[22]
By the 1960s–1970s quantum electrochemistry was developed by Revaz Dogonadze and his students.
21st century developments
In 2018, after ten years of research into electrochemical optimization techniques, Essam Elsahwi became the first to succeed in performing in-situ characterization of electrochemical stacks experiencing high industrial power levels, using the powerful technique of dielectric spectroscopy.[23]
2. Principles
Oxidation and reduction
The term "redox" stands for reduction-oxidation. It refers to electrochemical processes involving electron transfer to or from a molecule or ion, changing its oxidation state. This reaction can occur through the application of an external voltage or through the release of chemical energy. Oxidation and reduction describe the change of oxidation state that takes place in the atoms, ions or molecules involved in an electrochemical reaction. Formally, oxidation state is the hypothetical charge that an atom would have if all bonds to atoms of different elements were 100% ionic. An atom or ion that gives up an electron to another atom or ion has its oxidation state increase, and the recipient of the negatively charged electron has its oxidation state decrease.
For example, when atomic sodium reacts with atomic chlorine, sodium donates one electron and attains an oxidation state of +1. Chlorine accepts the electron and its oxidation state is reduced to −1. The sign of the oxidation state (positive/negative) actually corresponds to the value of each ion's electronic charge. The attraction of the differently charged sodium and chlorine ions is the reason they then form an ionic bond.
The loss of electrons from an atom or molecule is called oxidation, and the gain of electrons is reduction. This can be easily remembered through the use of mnemonic devices. Two of the most popular are "OIL RIG" (Oxidation Is Loss, Reduction Is Gain) and "LEO" the lion says "GER" (Lose Electrons: Oxidation, Gain Electrons: Reduction). Oxidation and reduction always occur in a paired fashion such that one species is oxidized when another is reduced. For cases where electrons are shared (covalent bonds) between atoms with large differences in electronegativity, the electron is assigned to the atom with the largest electronegativity in determining the oxidation state.
The atom or molecule which loses electrons is known as the reducing agent, or reductant, and the substance which accepts the electrons is called the oxidizing agent, or oxidant. Thus, the oxidizing agent is always being reduced in a reaction; the reducing agent is always being oxidized. Oxygen is a common oxidizing agent, but not the only one. Despite the name, an oxidation reaction does not necessarily need to involve oxygen. In fact, a fire can be fed by an oxidant other than oxygen; fluorine fires are often unquenchable, as fluorine is an even stronger oxidant (it has a weaker bond and higher electronegativity, and thus accepts electrons even better) than oxygen.
For reactions involving oxygen, the gain of oxygen implies the oxidation of the atom or molecule to which the oxygen is added (and the oxygen is reduced). In organic compounds, such as butane or ethanol, the loss of hydrogen implies oxidation of the molecule from which it is lost (and the hydrogen is reduced). This follows because the hydrogen donates its electron in covalent bonds with non-metals but it takes the electron along when it is lost. Conversely, loss of oxygen or gain of hydrogen implies reduction.
Balancing redox reactions
Electrochemical reactions in water are better analyzed by using the ion-electron method, where H+, OH− ion, H2O and electrons (to compensate the oxidation changes) are added to the cell's half-reactions for oxidation and reduction.
Acidic medium
In acidic medium, H+ ions and water are added to balance each half-reaction. For example, when manganese reacts with sodium bismuthate.
- Unbalanced reaction: Mn2+(aq) + NaBiO3(s) → Bi3+(aq) + MnO−4(aq)
- Oxidation: 4 H2O(l) + Mn2+(aq) → MnO−4(aq) + 8 H+(aq) + 5 e−
- Reduction: 2 e− + 6 H+(aq) + BiO−3(s) → Bi3+(aq) + 3 H2O(l)
Finally, the reaction is balanced by multiplying the stoichiometric coefficients so the numbers of electrons in both half reactions match
- 8 H2O(l) + 2 Mn2+(aq) → 2 MnO−4(aq) + 16 H+(aq) + 10 e−
- 10 e− + 30 H+(aq) + 5 BiO−3(s) → 5 Bi3+(aq) + 15 H2O(l)
and adding the resulting half reactions to give the balanced reaction:
- 14 H+(aq) + 2 Mn2+(aq) + 5 NaBiO3(s) → 7 H2O(l) + 2 MnO−4(aq) + 5 Bi3+(aq) + 5 Na+(aq)
Basic medium
In basic medium, OH− ions and water are added to balance each half-reaction. For example, in a reaction between potassium and sodium sulfite:
- Unbalanced reaction: KMnO4 + Na2SO3 + H2O → MnO2 + Na2SO4 + KOH
- Reduction: 3 e− + 2 H2O + MnO−4 → MnO2 + 4 OH−
- Oxidation: 2 OH− + SO2−3 → SO2−4 + H2O + 2 e−
Here, 'spectator ions' (K+, Na+) were omitted from the half-reactions. By multiplying the stoichiometric coefficients so the numbers of electrons in both half reaction match:
- 6 e− + 4 H2O + 2 MnO−4 → 2 MnO2 + 8 OH−
- 6 OH− + 3 SO2−3 → 3 SO2−4 + 3 H2O + 6 e−
the balanced overall reaction is obtained:
- 2 KMnO4 + 3 Na2SO3 + H2O → 2 MnO2 + 3 Na2SO4 + 2 KOH
Neutral medium
The same procedure as used in acidic medium can be applied, for example, to balance the complete combustion of propane:
- Unbalanced reaction: C3H8 + O2 → CO2 + H2O
- Reduction: 4 H+ + O2 + 4 e− → 2 H2O
- Oxidation: 6 H2O + C3H8 → 3 CO2 + 20 e− + 20 H+
By multiplying the stoichiometric coefficients so the numbers of electrons in both half reaction match:
- 20 H+ + 5 O2 + 20 e− → 10 H2O
- 6 H2O + C3H8 → 3 CO2 + 20 e− + 20 H+
the balanced equation is obtained:
- C3H8 + 5 O2 → 3 CO2 + 4 H2O
3. Electrochemical Cells
An electrochemical cell is a device that produces an electric current from energy released by a spontaneous redox reaction. This kind of cell includes the Galvanic cell or Voltaic cell, named after Luigi Galvani and Alessandro Volta, both scientists who conducted experiments on chemical reactions and electric current during the late 18th century.
Electrochemical cells have two conductive electrodes (the anode and the cathode). The anode is defined as the electrode where oxidation occurs and the cathode is the electrode where the reduction takes place. Electrodes can be made from any sufficiently conductive materials, such as metals, semiconductors, graphite, and even conductive polymers. In between these electrodes is the electrolyte, which contains ions that can freely move.
The galvanic cell uses two different metal electrodes, each in an electrolyte where the positively charged ions are the oxidized form of the electrode metal. One electrode will undergo oxidation (the anode) and the other will undergo reduction (the cathode). The metal of the anode will oxidize, going from an oxidation state of 0 (in the solid form) to a positive oxidation state and become an ion. At the cathode, the metal ion in solution will accept one or more electrons from the cathode and the ion's oxidation state is reduced to 0. This forms a solid metal that electrodeposits on the cathode. The two electrodes must be electrically connected to each other, allowing for a flow of electrons that leave the metal of the anode and flow through this connection to the ions at the surface of the cathode. This flow of electrons is an electric current that can be used to do work, such as turn a motor or power a light.
A galvanic cell whose electrodes are zinc and copper submerged in zinc sulfate and copper sulfate, respectively, is known as a Daniell cell.[24]
The half reactions in a Daniell cell are as follows:[24]
- Zinc electrode (anode): Zn(s) → Zn2+(aq) + 2 e−
- Copper electrode (cathode): Cu2+(aq) + 2 e− → Cu(s)

In this example, the anode is the zinc metal which is oxidized (loses electrons) to form zinc ions in solution, and copper ions accept electrons from the copper metal electrode and the ions deposit at the copper cathode as an electrodeposit. This cell forms a simple battery as it will spontaneously generate a flow of electric current from the anode to the cathode through the external connection. This reaction can be driven in reverse by applying a voltage, resulting in the deposition of zinc metal at the anode and formation of copper ions at the cathode.[24]
To provide a complete electric circuit, there must also be an ionic conduction path between the anode and cathode electrolytes in addition to the electron conduction path. The simplest ionic conduction path is to provide a liquid junction. To avoid mixing between the two electrolytes, the liquid junction can be provided through a porous plug that allows ion flow while minimizing electrolyte mixing. To further minimize mixing of the electrolytes, a salt bridge can be used which consists of an electrolyte saturated gel in an inverted U-tube. As the negatively charged electrons flow in one direction around this circuit, the positively charged metal ions flow in the opposite direction in the electrolyte.
A voltmeter is capable of measuring the change of electrical potential between the anode and the cathode.
The electrochemical cell voltage is also referred to as electromotive force or emf.
A cell diagram can be used to trace the path of the electrons in the electrochemical cell. For example, here is a cell diagram of a Daniell cell:
- Zn(s) | Zn2+ (1 M) || Cu2+ (1 M) | Cu(s)
First, the reduced form of the metal to be oxidized at the anode (Zn) is written. This is separated from its oxidized form by a vertical line, which represents the limit between the phases (oxidation changes). The double vertical lines represent the saline bridge on the cell. Finally, the oxidized form of the metal to be reduced at the cathode, is written, separated from its reduced form by the vertical line. The electrolyte concentration is given as it is an important variable in determining the exact cell potential.
4. Standard Electrode Potential
To allow prediction of the cell potential, tabulations of standard electrode potential are available. Such tabulations are referenced to the standard hydrogen electrode (SHE). The standard hydrogen electrode undergoes the reaction
- 2 H+(aq) + 2 e− → H2
which is shown as a reduction but, in fact, the SHE can act as either the anode or the cathode, depending on the relative oxidation/reduction potential of the other electrode/electrolyte combination. The term standard in SHE requires a supply of hydrogen gas bubbled through the electrolyte at a pressure of 1 atm and an acidic electrolyte with H+ activity equal to 1 (usually assumed to be [H+] = 1 mol/liter, i.e. pH = 0).
The SHE electrode can be connected to any other electrode by a salt bridge and an external circuit to form a cell. If the second electrode is also at standard conditions, then the measured cell potential is called the standard electrode potential for the electrode. The standard electrode potential for the SHE is zero, by definition. The polarity of the standard electrode potential provides information about the relative reduction potential of the electrode compared to the SHE. If the electrode has a positive potential with respect to the SHE, then that means it is a strongly reducing electrode which forces the SHE to be the anode (an example is Cu in aqueous CuSO4 with a standard electrode potential of 0.337 V). Conversely, if the measured potential is negative, the electrode is more oxidizing than the SHE (such as Zn in ZnSO4 where the standard electrode potential is −0.76 V).[24]
Standard electrode potentials are usually tabulated as reduction potentials. However, the reactions are reversible and the role of a particular electrode in a cell depends on the relative oxidation/reduction potential of both electrodes. The oxidation potential for a particular electrode is just the negative of the reduction potential. A standard cell potential can be determined by looking up the standard electrode potentials for both electrodes (sometimes called half cell potentials). The one that is smaller will be the anode and will undergo oxidation. The cell potential is then calculated as the sum of the reduction potential for the cathode and the oxidation potential for the anode.
- E°cell = E°red (cathode) – E°red (anode) = E°red (cathode) + E°oxi (anode)
For example, the standard electrode potential for a copper electrode is:
Cell diagram
- Pt(s) | H2 (1 atm) | H+ (1 M) || Cu2+ (1 M) | Cu(s)
- E°cell = E°red (cathode) – E°red (anode)
At standard temperature, pressure and concentration conditions, the cell's emf (measured by a multimeter) is 0.34 V. By definition, the electrode potential for the SHE is zero. Thus, the Cu is the cathode and the SHE is the anode giving
- Ecell = E°(Cu2+/Cu) – E°(H+/H2)
Or,
- E°(Cu2+/Cu) = 0.34 V
Changes in the stoichiometric coefficients of a balanced cell equation will not change the E°red value because the standard electrode potential is an intensive property.
5. Spontaneity of Redox Reaction
During operation of an electrochemical cell, chemical energy is transformed into electrical energy. This can be expressed mathematically as the product of the cell's emf Ecell measured in volts (V) and the electric charge Qele,trans transferred through the external circuit.
- Electrical energy = EcellQele,trans
Qele,trans is the cell current integrated over time and measured in coulombs (C); it can also be determined by multiplying the total number ne of electrons transferred (measured in moles) times Faraday's constant (F).
The emf of the cell at zero current is the maximum possible emf. It can be used to calculate the maximum possible electrical energy that could be obtained from a chemical reaction. This energy is referred to as electrical work and is expressed by the following equation:
- [math]\displaystyle{ W_\mathrm{max} = W_\mathrm{electrical} = -n_eFE_\mathrm{cell} }[/math],
where work is defined as positive when it increases the energy of the system.
Since the free energy is the maximum amount of work that can be extracted from a system, one can write:[25]
- [math]\displaystyle{ \Delta G = -n_eFE_\mathrm{cell} }[/math]
A positive cell potential gives a negative change in Gibbs free energy. This is consistent with the cell production of an electric current from the cathode to the anode through the external circuit. If the current is driven in the opposite direction by imposing an external potential, then work is done on the cell to drive electrolysis.[25]
A spontaneous electrochemical reaction (change in Gibbs free energy less than zero) can be used to generate an electric current in electrochemical cells. This is the basis of all batteries and fuel cells. For example, gaseous oxygen (O2) and hydrogen (H2) can be combined in a fuel cell to form water and energy, typically a combination of heat and electrical energy.[25]
Conversely, non-spontaneous electrochemical reactions can be driven forward by the application of a current at sufficient voltage. The electrolysis of water into gaseous oxygen and hydrogen is a typical example.
The relation between the equilibrium constant, K, and the Gibbs free energy for an electrochemical cell is expressed as follows:
- [math]\displaystyle{ \Delta G^\circ = -RT \ln K = -nFE^{\circ}_\mathrm{cell} }[/math].
Rearranging to express the relation between standard potential and equilibrium constant yields
- [math]\displaystyle{ E^{\circ}_{cell} = \frac{RT}{nF}\ln K }[/math].
At T = 298 K, the previous equation can be rewritten using the Briggsian logarithm as follows:
- [math]\displaystyle{ E^{\circ}_{cell} = \frac{0.05916\,\mathrm{V}}{n} \log K }[/math]
6. Cell Emf Dependency on Changes in Concentration
6.1. Nernst Equation
The standard potential of an electrochemical cell requires standard conditions (ΔG°) for all of the reactants. When reactant concentrations differ from standard conditions, the cell potential will deviate from the standard potential. In the 20th century German chemist Walther Nernst proposed a mathematical model to determine the effect of reactant concentration on electrochemical cell potential.
In the late 19th century, Josiah Willard Gibbs had formulated a theory to predict whether a chemical reaction is spontaneous based on the free energy
- [math]\displaystyle{ \Delta G = \Delta G^\circ + RT \ln Q }[/math]
Here ΔG is change in Gibbs free energy, ΔG° is the cell potential when Q is equal to 1, T is absolute temperature (Kelvin), R is the gas constant and Q is the reaction quotient, which can be calculated by dividing concentrations of products by those of reactants, each raised to the power of its stoichiometric coefficient, using only those products and reactants that are aqueous or gaseous.
Gibbs' key contribution was to formalize the understanding of the effect of reactant concentration on spontaneity.
Based on Gibbs' work, Nernst extended the theory to include the contribution from electric potential on charged species. As shown in the previous section, the change in Gibbs free energy for an electrochemical cell can be related to the cell potential. Thus, Gibbs' theory becomes
- [math]\displaystyle{ n_eF\Delta E = n_e F\Delta E^\circ - RT \ln Q }[/math]
Here ne is the number of electrons (in moles), F is the Faraday constant (in coulombs/mole), and ΔE is the cell potential (in volts).
Finally, Nernst divided through by the amount of charge transferred to arrive at a new equation which now bears his name:
- [math]\displaystyle{ \Delta E = \Delta E^\circ - \frac{RT}{nF} \ln Q }[/math]
Assuming standard conditions (T = 298 K or 25 °C) and R = 8.3145 J/(K·mol), the equation above can be expressed on base—10 logarithm as shown below:[26]
- [math]\displaystyle{ \Delta E = \Delta E^\circ- \frac{0.05916 \,\mathrm{V}}{n} \log Q }[/math]
Note that RT/F is also known as the thermal voltage VT and is found in the study of plasmas and semiconductors as well. The value 0.05916 V in the above equation is just the thermal voltage at standard temperature multiplied by the natural logarithm of 10.
6.2. Concentration Cells
A concentration cell is an electrochemical cell where the two electrodes are the same material, the electrolytes on the two half-cells involve the same ions, but the electrolyte concentration differs between the two half-cells.
An example is an electrochemical cell, where two copper electrodes are submerged in two copper(II) sulfate solutions, whose concentrations are 0.05 M and 2.0 M, connected through a salt bridge. This type of cell will generate a potential that can be predicted by the Nernst equation. Both can undergo the same chemistry (although the reaction proceeds in reverse at the anode)
- Cu2+(aq) + 2 e− → Cu(s)
Le Chatelier's principle indicates that the reaction is more favorable to reduction as the concentration of Cu2+ ions increases. Reduction will take place in the cell's compartment where the concentration is higher and oxidation will occur on the more dilute side.
The following cell diagram describes the concentration cell mentioned above:
- Cu(s) | Cu2+ (0.05 M) || Cu2+ (2.0 M) | Cu(s)
where the half cell reactions for oxidation and reduction are:
- Oxidation: Cu(s) → Cu2+ (0.05 M) + 2 e−
- Reduction: Cu2+ (2.0 M) + 2 e− → Cu(s)
- Overall reaction: Cu2+ (2.0 M) → Cu2+ (0.05 M)
The cell's emf is calculated through the Nernst equation as follows:
- [math]\displaystyle{ E = E^\circ - \frac{0.05916 \,\mathrm{V}}{2} \log \frac{[\mathrm{Cu^{2+}}]_\mathrm{diluted}}{[\mathrm{Cu^{2+}}]_\mathrm{concentrated}} }[/math]
The value of E° in this kind of cell is zero, as electrodes and ions are the same in both half-cells.
After replacing values from the case mentioned, it is possible to calculate cell's potential:
- [math]\displaystyle{ E = 0 - \frac{0.05916 \,\mathrm{V}}{2} \log \frac{0.05}{2.0} = 0.0474\,\mathrm{V} }[/math]
or by:
- [math]\displaystyle{ E = 0 - \frac{0.0257 \,\mathrm{V}}{2} \ln \frac{0.05}{2.0}= 0.0474\,\mathrm{V} }[/math]
However, this value is only approximate, as reaction quotient is defined in terms of ion activities which can be approximated with the concentrations as calculated here.
The Nernst equation plays an important role in understanding electrical effects in cells and organelles. Such effects include nerve synapses and cardiac beat as well as the resting potential of a somatic cell.
7. Battery
Many types of battery have been commercialized and represent an important practical application of electrochemistry.[27] Early wet cells powered the first telegraph and telephone systems, and were the source of current for electroplating. The zinc-manganese dioxide dry cell was the first portable, non-spillable battery type that made flashlights and other portable devices practical. The mercury battery using zinc and mercuric oxide provided higher levels of power and capacity than the original dry cell for early electronic devices, but has been phased out of common use due to the danger of mercury pollution from discarded cells.
The lead–acid battery was the first practical secondary (rechargeable) battery that could have its capacity replenished from an external source. The electrochemical reaction that produced current was (to a useful degree) reversible, allowing electrical energy and chemical energy to be interchanged as needed. Common lead acid batteries contain a mixture of sulfuric acid and water, as well as lead plates. The most common mixture used today is 30% acid. One problem, however, is if left uncharged acid will crystallize within the lead plates of the battery rendering it useless. These batteries last an average of 3 years with daily use but it is not unheard of for a lead acid battery to still be functional after 7–10 years. Lead-acid cells continue to be widely used in automobiles.
All the preceding types have water-based electrolytes, which limits the maximum voltage per cell. The freezing of water limits low temperature performance. The lithium battery, which does not (and cannot) use water in the electrolyte, provides improved performance over other types; a rechargeable lithium-ion battery is an essential part of many mobile devices.
The flow battery, an experimental type, offers the option of vastly larger energy capacity because its reactants can be replenished from external reservoirs. The fuel cell can turn the chemical energy bound in hydrocarbon gases or hydrogen and oxygen directly into electrical energy with a much higher efficiency than any combustion process; such devices have powered many spacecraft and are being applied to grid energy storage for the public power system.
8. Corrosion
Corrosion is an electrochemical process, which reveals itself as rust or tarnish on metals like iron or copper and their respective alloys, steel and brass.
8.1. Iron Corrosion
For iron rust to occur the metal has to be in contact with oxygen and water. The chemical reactions for this process are relatively complex and not all of them are completely understood. It is believed the causes are the following: Electron transfer (reduction-oxidation)
- One area on the surface of the metal acts as the anode, which is where the oxidation (corrosion) occurs. At the anode, the metal gives up electrons.
- Fe(s) → Fe2+(aq) + 2 e−
- Electrons are transferred from iron, reducing oxygen in the atmosphere into water on the cathode, which is placed in another region of the metal.
- O2(g) + 4 H+(aq) + 4 e− → 2 H2O(l)
- Global reaction for the process:
- 2 Fe(s) + O2(g) + 4 H+(aq) → 2 Fe2+(aq) + 2 H2O(l)
- Standard emf for iron rusting:
- E° = E° (cathode) − E° (anode)
- E° = 1.23V − (−0.44 V) = 1.67 V
Iron corrosion takes place in an acid medium; H+ ions come from reaction between carbon dioxide in the atmosphere and water, forming carbonic acid. Fe2+ ions oxidize further, following this equation:
- 4 Fe2+(aq) + O2(g) + (4+2x) H2O(l) → 2 Fe2O3·xH2O + 8 H+(aq)
Iron(III) oxide hydrate is known as rust. The concentration of water associated with iron oxide varies, thus the chemical formula is represented by Fe2O3·xH2O.
An electric circuit is formed as passage of electrons and ions occurs; thus if an electrolyte is present it will facilitate oxidation, explaining why rusting is quicker in salt water.
8.2. Corrosion of Common Metals
Coinage metals, such as copper and silver, slowly corrode through use. A patina of green-blue copper carbonate forms on the surface of copper with exposure to the water and carbon dioxide in the air. Silver coins or cutlery that are exposed to high sulfur foods such as eggs or the low levels of sulfur species in the air develop a layer of black silver sulfide.
Gold and platinum are extremely difficult to oxidize under normal circumstances, and require exposure to a powerful chemical oxidizing agent such as aqua regia.
Some common metals oxidize extremely rapidly in air. Titanium and aluminium oxidize instantaneously in contact with the oxygen in the air. These metals form an extremely thin layer of oxidized metal on the surface, which bonds with the underlying metal. This thin oxide layer protects the underlying bulk of the metal from the air preventing the entire metal from oxidizing. These metals are used in applications where corrosion resistance is important. Iron, in contrast, has an oxide that forms in air and water, called rust, that does not bond with the iron and therefore does not stop the further oxidation of the iron. Thus iron left exposed to air and water will continue to rust until all of the iron is oxidized.
8.3. Prevention of Corrosion
Attempts to save a metal from becoming anodic are of two general types. Anodic regions dissolve and destroy the structural integrity of the metal.
While it is almost impossible to prevent anode/cathode formation, if a non-conducting material covers the metal, contact with the electrolyte is not possible and corrosion will not occur.
Coating
Metals can be coated with paint or other less conductive metals (passivation). This prevents the metal surface from being exposed to electrolytes. Scratches exposing the metal substrate will result in corrosion. The region under the coating adjacent to the scratch acts as the anode of the reaction.
See Anodizing
Sacrificial anodes
A method commonly used to protect a structural metal is to attach a metal which is more anodic than the metal to be protected. This forces the structural metal to be cathodic, thus spared corrosion. It is called "sacrificial" because the anode dissolves and has to be replaced periodically.
Zinc bars are attached to various locations on steel ship hulls to render the ship hull cathodic. The zinc bars are replaced periodically. Other metals, such as magnesium, would work very well but zinc is the least expensive useful metal.
To protect pipelines, an ingot of buried or exposed magnesium (or zinc) is buried beside the pipeline and is connected electrically to the pipe above ground. The pipeline is forced to be a cathode and is protected from being oxidized and rusting. The magnesium anode is sacrificed. At intervals new ingots are buried to replace those dissolved.
9. Electrolysis
The spontaneous redox reactions of a conventional battery produce electricity through the different reduction potentials of the cathode and anode in the electrolyte. However, electrolysis requires an external source of electrical energy to induce a chemical reaction, and this process takes place in a compartment called an electrolytic cell.
9.1. Electrolysis of Molten Sodium Chloride
When molten, the salt sodium chloride can be electrolyzed to yield metallic sodium and gaseous chlorine. Industrially this process takes place in a special cell named Down's cell. The cell is connected to an electrical power supply, allowing electrons to migrate from the power supply to the electrolytic cell.[28]
Reactions that take place in a Down's cell are the following:[28]
- Anode (oxidation): 2 Cl− → Cl2(g) + 2 e−
- Cathode (reduction): 2 Na+(l) + 2 e− → 2 Na(l)
- Overall reaction: 2 Na+ + 2 Cl−(l) → 2 Na(l) + Cl2(g)
This process can yield large amounts of metallic sodium and gaseous chlorine, and is widely used in mineral dressing and metallurgy industries.
The emf for this process is approximately −4 V indicating a (very) non-spontaneous process. In order for this reaction to occur the power supply should provide at least a potential difference of 4 V. However, larger voltages must be used for this reaction to occur at a high rate.
9.2. Electrolysis of Water
Water can be converted to its component elemental gases, H2 and O2, through the application of an external voltage. Water does not decompose into hydrogen and oxygen spontaneously as the Gibbs free energy change for the process at standard conditions is very positive, about 474.4 kJ. The decomposition of water into hydrogen and oxygen can be performed in an electrolytic cell. In it, a pair of inert electrodes usually made of platinum immersed in water act as anode and cathode in the electrolytic process. The electrolysis starts with the application of an external voltage between the electrodes. This process will not occur except at extremely high voltages without an electrolyte such as sodium chloride or sulfuric acid (most used 0.1 M).[29]
Bubbles from the gases will be seen near both electrodes. The following half reactions describe the process mentioned above:
- Anode (oxidation): 2 H2O(l) → O2(g) + 4 H+(aq) + 4 e−
- Cathode (reduction): 2 H2O(g) + 2 e− → H2(g) + 2 OH−(aq)
- Overall reaction: 2 H2O(l) → 2 H2(g) + O2(g)
Although strong acids may be used in the apparatus, the reaction will not net consume the acid. While this reaction will work at any conductive electrode at a sufficiently large potential, platinum catalyzes both hydrogen and oxygen formation, allowing for relatively low voltages (~2 V depending on the pH).[29]
9.3. Electrolysis of Aqueous Solutions
Electrolysis in an aqueous solution is a similar process as mentioned in electrolysis of water. However, it is considered to be a complex process because the contents in solution have to be analyzed in half reactions, whether reduced or oxidized.
9.4. Electrolysis of a Solution of Sodium Chloride
The presence of water in a solution of sodium chloride must be examined in respect to its reduction and oxidation in both electrodes. Usually, water is electrolysed as mentioned above in electrolysis of water yielding gaseous oxygen in the anode and gaseous hydrogen in the cathode. On the other hand, sodium chloride in water dissociates in Na+ and Cl− ions. The cation, which is the positive ion, will be attracted to the cathode (−), thus reducing the sodium ion. The chloride anion will then be attracted to the anode (+), where it is oxidized to chlorine gas.[30]
The following half reactions should be considered in the process mentioned:[30]
- Cathode: Na+(aq) + e− → Na(s) E°red = –2.71 V
- Anode: 2 Cl−(aq) → Cl2(g) + 2 e− E°red = +1.36 V
- Cathode: 2 H2O(l) + 2 e− → H2(g) + 2 OH−(aq) E°red = –0.83 V
- Anode: 2 H2O(l) → O2(g) + 4 H+(aq) + 4 e− E°red = +1.23 V
Reaction 1 is discarded as it has the most negative value on standard reduction potential thus making it less thermodynamically favorable in the process.
When comparing the reduction potentials in reactions 2 and 4, the reduction of chloride ion is favored. Thus, if the Cl− ion is favored for reduction, then the water reaction is favored for oxidation producing gaseous oxygen, however experiments show gaseous chlorine is produced and not oxygen.
Although the initial analysis is correct, there is another effect, known as the overvoltage effect. Additional voltage is sometimes required, beyond the voltage predicted by the E°cell. This may be due to kinetic rather than thermodynamic considerations. In fact, it has been proven that the activation energy for the chloride ion is very low, hence favorable in kinetic terms. In other words, although the voltage applied is thermodynamically sufficient to drive electrolysis, the rate is so slow that to make the process proceed in a reasonable time frame, the voltage of the external source has to be increased (hence, overvoltage).[30]
Finally, reaction 3 is favorable because it describes the proliferation of OH− ions thus letting a probable reduction of H+ ions less favorable an option.
The overall reaction for the process according to the analysis is the following:[30]
- Anode (oxidation): 2 Cl−(aq) → Cl2(g) + 2 e−
- Cathode (reduction): 2 H2O(l) + 2 e− → H2(g) + 2 OH−(aq)
- Overall reaction: 2 H2O + 2 Cl−(aq) → H2(g) + Cl2(g) + 2 OH−(aq)
As the overall reaction indicates, the concentration of chloride ions is reduced in comparison to OH− ions (whose concentration increases). The reaction also shows the production of gaseous hydrogen, chlorine and aqueous sodium hydroxide.
Quantitative electrolysis and Faraday's laws
Quantitative aspects of electrolysis were originally developed by Michael Faraday in 1834. Faraday is also credited to have coined the terms electrolyte, electrolysis, among many others while he studied quantitative analysis of electrochemical reactions. Also he was an advocate of the law of conservation of energy.
First law
Faraday concluded after several experiments on electric current in a non-spontaneous process that the mass of the products yielded on the electrodes was proportional to the value of current supplied to the cell, the length of time the current existed, and the molar mass of the substance analyzed. In other words, the amount of a substance deposited on each electrode of an electrolytic cell is directly proportional to the quantity of electricity passed through the cell.[31]
Below is a simplified equation of Faraday's first law:
- [math]\displaystyle{ m = \frac{1}{96485 \mathrm{(C\cdot mol^{-1})}} \cdot \frac{QM}{n} }[/math]
where
- m is the mass of the substance produced at the electrode (in grams),
- Q is the total electric charge that passed through the solution (in coulombs),
- n is the valence number of the substance as an ion in solution (electrons per ion),
- M is the molar mass of the substance (in grams per mole).
Second law
Faraday devised the laws of chemical electrodeposition of metals from solutions in 1857. He formulated the second law of electrolysis stating "the amounts of bodies which are equivalent to each other in their ordinary chemical action have equal quantities of electricity naturally associated with them." In other words, the quantities of different elements deposited by a given amount of electricity are in the ratio of their chemical equivalent weights.[32]
An important aspect of the second law of electrolysis is electroplating, which together with the first law of electrolysis has a significant number of applications in industry, as when used to protectively coat metals to avoid corrosion.
10. Applications
There are various extremely important electrochemical processes in both nature and industry, like the coating[33][34] of objects with metals or metal oxides through electrodeposition, the addition (electroplating) or removal (electropolishing) of thin layers of metal from an object's surface,[35] and the detection of alcohol in drunk drivers through the redox reaction of ethanol. The generation of chemical energy through photosynthesis is inherently an electrochemical process, as is production of metals like aluminum and titanium from their ores. Certain diabetes blood sugar meters measure the amount of glucose in the blood through its redox potential. In addition to established electrochemical technologies (like deep cycle lead acid batteries) there is also a wide range of new emerging technologies such as fuel cells, large format lithium-ion batteries, electrochemical reactors and super-capacitors that are becoming increasingly commercial.[36] Electrochemistry also has important applications in the food industry, like the assessment of food/package interactions,[37] the analysis of milk composition,[38] the characterization and the determination of the freezing end-point of ice-cream mixes, or the determination of free acidity in olive oil.
References
- Richard P. Olenick, Tom M. Apostol, David L. Goodstein Beyond the mechanical universe: from electricity to modern physics, Cambridge University Press (1986) ISBN:0-521-30430-X, p. 160 https://books.google.com/books?id=Ht4T7C7AXZIC&pg=PA160
- R. Hellborg Electrostatic accelerators: fundamentals and applications (2005) ISBN:3540239839 p. 52 https://books.google.com/books?id=tc6CEuIV1jEC&pg=PA52
- Steven Weinberg The discovery of subatomic particles Cambridge University Press (2003) ISBN:0-521-82351-X, p. 15 https://books.google.com/books?id=cKXuMfnMC4IC&pg=PA15
- J. A. M. Bleeker, Johannes Geiss, M. Huber The century of space science, Volume 1, Springer (2001) ISBN:0-7923-7196-8 p. 227 https://books.google.com/books?id=NMk3adgqfawC&pg=PA227
- John Robert Norris, Douglas W. Ribbons (1972) Methods in microbiology, Volume 6, Academic Press. ISBN:0-12-521546-0 p. 248 https://books.google.com/books?id=TFfQPQKSc3EC&pg=PA248
- Frederick Collier Bakewell Electric science; its history, phenomena, and applications, Ingram, Cooke (1853) pp. 27–31 https://archive.org/details/bub_gb_Lks1AAAAMAAJ/page/n32
- Keith James Laidler The world of physical chemistry, Oxford University Press (1995) ISBN:0-19-855919-4 pp. 219–220 https://books.google.com/books?id=01LRlPbH80cC&pg=PA219
- The New Encyclopædia Britannica: Micropædia, Vol. 10 (1991) ISBN:0-85229-529-4, p. 90
- Charles Knight (ed.) Biography: or, Third division of "The English encyclopedia", Volume 2, Bradbury, Evans & Co. (1867) https://books.google.com/books?id=BchPAAAAMAAJ&pg=RA2-PT168
- William Berkson (1974) Fields of force: the development of a world view from Faraday to Einstein, Routledge. ISBN:0-7100-7626-6 pp. 34 ff https://archive.org/details/fieldsofforcedev0000berk/page/34
- Brian Scott Baigrie Electricity and magnetism: a historical perspective, Greenwood Publishing Group (2007) ISBN:0-313-33358-0 p. 73 https://books.google.com/books?id=3XEc5xkWxi4C&pg=PA73
- Nobel Lectures, p. 59
- Polmear, I.J. (2006). "Production of Aluminium". Light alloys from traditional alloys to nanocrystals. Oxford: Elsevier/Butterworth-Heinemann. pp. 15–16. ISBN 978-0-7506-6371-7. https://books.google.com/books?id=td0jD4it63cC&pg=PT29.
- Nobel Lectures, p. 170
- Nobel Lectures, p. 363
- Nobel Lectures, p. 342
- "About ECS". https://www.electrochem.org/about/.
- Millikan, Robert A. (1911). "The Isolation of an Ion, a Precision Measurement of its Charge, and the Correction of Stokes' Law". Physical Review 32 (2): 349–397. doi:10.1063/1.2743125. Bibcode: 1911PhRvI..32..349M. https://authors.library.caltech.edu/6437/1/MILpr11b.pdf.
- "Remembering the oil-drop experiment". Physics Today 60 (5, 56). 2007. doi:10.1063/1.2743125. https://dx.doi.org/10.1063%2F1.2743125
- William L. Masterton, Cecile N. Hurley Chemistry: Principles and Reactions, Cengage Learning (2008) ISBN:0-495-12671-3 p. 379 https://books.google.com/books?id=teubNK-b2bsC&pg=PT379
- The Nobel Prize in Chemistry 1948 Arne Tiselius, nobelprize.org http://nobelprize.org/nobel_prizes/chemistry/laureates/1948/tiselius-bio.html
- The International Society of Electrochemistry http://www.ise-online.org/geninfo/index.php
- "Pulsenics-03". https://www.pulsenics.com/.
- Wiberg, pp. 215–216
- Swaddle, pp. 308–314
- Wiberg, pp. 210–212
- Badwal, Sukhvinder P. S.; Giddey, Sarbjit S.; Munnings, Christopher; Bhatt, Anand I.; Hollenkamp, Anthony F. (24 September 2014). "Emerging electrochemical energy conversion and storage technologies (open access)". Frontiers in Chemistry 2: 79. doi:10.3389/fchem.2014.00079. PMID 25309898. Bibcode: 2014FrCh....2...79B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4174133
- Ebbing, pp. 800–801
- Wiberg, pp. 235–239
- Ebbing, pp. 837–839
- Wiberg, p. 65
- Faraday, Michael (1791–1867), Wolfram Research http://scienceworld.wolfram.com/biography/Faraday.html
- McIntyre, J.M.; Pham, H.Q. (1996). "Electrochemical impedance spectroscopy; a tool for organic coatings optimizations". Progress in Organic Coatings 27 (1–4): 201–207. doi:10.1016/0300-9440(95)00532-3. https://dx.doi.org/10.1016%2F0300-9440%2895%2900532-3
- Amirudin, A.; Thieny, D. (1995). "Application of electrochemical impedance spectroscopy to study the degradation of polymer-coated metals". Progress in Organic Coatings 26 (1): 1–28. doi:10.1016/0300-9440(95)00581-1. https://dx.doi.org/10.1016%2F0300-9440%2895%2900581-1
- "What is Electropolishing?" https://www.electro-glo.com/what-is-electropolishing/
- S.P.S BADWAL; S. Giddey; C. Munnings; A. I. Bhatt; A. Hollenkamp (2014). "Emerging electrochemical energy conversion and storage technologies". Frontiers in Chemistry 2: 79. doi:10.3389/fchem.2014.00079. PMID 25309898. Bibcode: 2014FrCh....2...79B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4174133
- Hollaender, J. (2009). "Rapid assessment of food/package interactions by electrochemical impedance spectroscopy (EIS)". Food Additives & Contaminants 14 (6–7): 617–626. doi:10.1080/02652039709374574. PMID 9373526. https://dx.doi.org/10.1080%2F02652039709374574
- Mabrook, M.F.; Petty, M.C. (2003). "Effect of composition on the electrical conductance of milk". Journal of Food Engineering 60 (3): 321–325. doi:10.1016/S0260-8774(03)00054-2. https://dx.doi.org/10.1016%2FS0260-8774%2803%2900054-2


