 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chiao-En Wu | + 1965 word(s) | 1965 | 2020-11-05 12:33:18 | | | |
| 2 | Peter Tang | Meta information modification | 1965 | 2020-11-06 06:48:48 | | |
Video Upload Options
Gemcitabine-based chemotherapy is the current standard treatment for biliary tract cancers (BTCs) and resistance to gemcitabine remains the clinical challenge. TP53 mutation has been shown to be associated with poor clinicopathologic characteristics and survival in patients with BTCs, indicating that p53 plays an important role in the treatment of these cancers.
1. Current Treatment for Biliary Tract Cancers (BTCs): Chemotherapy, Targeted Therapy, and Immunotherapy
Biliary tract cancers (BTCs) including intrahepatic cholangiocarcinoma (iCCA), extrahepatic cholangiocarcinoma (eCCA), gallbladder cancer, and ampullary cancer are a group of relatively rare cancers arising from the epithelium of the biliary tract [1][2][3][4] and have aggressive biological behavior, as they are diagnosed at an advanced stage with poor prognosis and have a high recurrence rate after primary surgery [5][6]. Gemcitabine and cisplatin have been the standard treatments in first-line chemotherapy since the ABC-02 trial was published in 2010 [7]. Clinical trials have evaluated molecular targeted therapies in combination with chemotherapy; however, none of the completed phase III trials [8][9][10][11] and phase II studies [12][13][14][15] have demonstrated significant improvement in progression-free survival (PFS) and overall survival (OS) in patients with advanced BTCs [16]. Real word experience has confirmed the feasibility and safety of gemcitabine and cisplatin in advanced BTCs [6][17].
With the development of comprehensive genetic profiling by next generation sequencing (NGS), alterations of isocitrate dehydrogenase genes (IDH1, IDH2) and fibroblast growth factor receptors (FGFR1, FGFR2, FGFR3) were found in iCCA and targeting such genetic alterations has become a novel therapeutic strategy in iCCA [18][19]. Recently, pemigatinib (Pemazyre), a FGFR inhibitor, has demonstrated activity in previously treated, unresectable locally advanced or metastatic CCA with FGFR2 fusions or rearrangements [20]. Such FGFR2 alternations account for 15% of iCCA [19] and are not found in other BTCs, limiting its general application in BTCs. In addition, pemigatinib is approved to be used for chemotherapy-treated CCA patients, so chemotherapy is still the first-line standard treatment for iCCA patients.
Although immune checkpoint inhibitors (ICIs) showed limited efficacy in unselected patents [21], a number of biomarker-guided immunotherapy studies are currently recruiting for ongoing trials [22]. Pembrolizumab was approved for treatment of a variety of advanced microsatellite instability-high (MSI-H) or deficient mismatch repair (dMMR) solid tumors (including BTC) [23] in 2017, but MSI-H or dMMR rarely occur in BTCs, including iCCA [24][25]. Recently, pembrolizumab was approved for treatment of tumor mutation burden-high (TMB-H) solid tumors, which is defined as ≥ 10 mutations/megabase (mut/Mb) assessed by FoundationOneCDx assay. Similar to MSI-H/dMMR, TMB-H rarely happens in BTCs, limiting the utility of ICIs in BTCs. Combination trials of ICIs and chemotherapy are ongoing to explore the potential for future application in BTCs.
Therefore, chemotherapy is still the mainstay of treatment in advanced BTCs. In CCA patients with FGFR2 fusion or rearrangement, pemigatinib is a late-line treatment option. The use of ICIs such as pembrolizumab is limited to CCA patients with MSI-H/dMMR or TMB-H.
2. Introduction to p53 in Cancer
In 1979, p53 was originally discovered by several groups as the host tumor protein targeted by the large tumor antigen of SV40 DNA tumor virus [26][27][28][29][30]. The TP53 gene, which encodes p53, was generally considered to be an "oncogene" before the mid-1980s [31][32][33], when sequencing of p53 cDNA clones revealed that some of them carried mutations that were comparable to the sequence of murine wild-type p53 derived from normal tissues [34][35]. It became clear that p53 mutations are frequently present in tumor-derived cell lines [34] and it was found that mouse tumor derived mutant (MUT) p53 could indeed promote cell transformation and survival but wild-type (WT) p53 did not. Deletion mapping studies accompanied by DNA sequencing demonstrated a frequent two-hit p53 allele inactivation mechanism in human tumors and strongly indicated that p53 works as a tumor suppressor [36]. The observation that transformation of cultured cells could be repressed by overexpression of WT p53 confirmed its tumor suppressor role [37]. Further confirmation of the tumor suppressor role of p53 and the two-hit inactivation mechanism came from the demonstration of mono-allelic germline TP53 mutations in Li-Fraumeni familial cancer predisposition syndrome, and the second hit inactivation was evident in the tumors of the affected Li-Fraumeni patients [38].
In the early 1990s, p53 was found to be a transcription factor [39] that can bind tightly to a specific DNA consensus sequence [40]. The abilities to bind to specific sequences and transactivate particular genes distinguishes WT p53 from all cancer-associated p53 mutants. CDKN1A encoding the cyclin-dependent kinase inhibitor p21 [41] and the proapoptotic BAX gene [42] were found to be transactivated by p53. Subsequently, a number of additional p53 transcriptionally targeted genes have been identified, most of which encode proteins that are intimately involved in apoptosis or in control of cell cycle progression. In addition, two of the transcriptionally targeted genes, MDM2 and PPM1D, have an autoregulatory feedback role to regulate p53.
The MDM2 protein, the most important protein directly interacting with p53, was discovered in 1992. MDM2, an E3 ubiquitin ligase, was shown to bind to p53 tightly and inhibit p53 transcriptional activity [43] as well as promoting the ubiquitylation and subsequent proteasomal degradation of p53 [44]. MDMX (MDM4 in the mouse) was discovered as a paralogue of MDM2 in 1996, which can form heterodimers with MDM2 to augment MDM2 activity and contribute to p53 degradation but has no measurable intrinsic E3 ligase activity [45][46]. The intimate relationship between MDM2 and p53 was demonstrated by gene knockout studies in mice. Knockout of the MDM2 gene was found to be embryonically lethal but could be rescued by additional knockout of the mouse p53 gene [47].
Activation of p53 occurs in response to cellular stress, particularly as a consequence of DNA damage response (DDR), such as induced by gemcitabine. The DDR signaling results in phosphorylation of p53 and MDM2, which prevents MDM2 from binding to p53. This allows levels of the p53 protein to increase and activates its function as a transcription factor [48][49] to drive the expression of p53 target genes, which execute the appropriate cellular responses, including DNA repair, altered metabolism, cell cycle arrest, or apoptosis and senescence [50]. The TP53 gene is one of the most frequently mutated genes in human cancer [51] and has been found to be either mutated or the p53 protein functionally inactivated in most human cancers [52][53]. Homozygous mutation and/or deletion of TP53 results in loss of WT p53 tumor suppressor function. However, some of the point missense mutations have also been shown to have dominant oncogenic functions (gain-of-function mutations) that can override WT p53 from a remaining WT TP53 allele, via binding to other transcription factors that transactivate genes associated with tumor survival and drug resistance [54] (Figure 1).

Figure 1. The influence of TP53 mutation in cancer cells. p53 is a transcription factor that transactivates downstream genes responsible for cell cycle arrest, apoptosis and cell senescence. Once p53 becomes mutated (MUT), it loses wild-type (WT) p53 function. In some cases, the mutant form selectively accumulates due to loss of binding to MDM2. As p53 functions as a tetramer, the mutant and functionally defective form then dominates over the lower number of wild-type p53 protein molecules from the remaining normal allele. However, for the most part, TP53 behaves as a classical tumor suppressor gene and both alleles need to be inactivated by a combination of mutation and/or deletion. Nevertheless, some mutant forms of p53 bind to transcription factors (TFs), which transactivate genes responsible for tumor survival and drug resistance. Taken together, MUT p53 enhances tumor growth.
3. p53 Plays a Role in Gemcitabine Resistance in Cancers
Gemcitabine treatment induces a cellular DNA damage response (DDR) signaling cascade involving activation by phosphorylation of ataxia telangiectasia mutated (ATM), ataxia telangiectasia related (ATR) and p53. Once p53WT is activated by phosphorylation and subsequent accumulation, it induces cell cycle arrest, allowing and facilitating DNA repair, or, depending on the cell type and level of damage, it triggers cell death (by apoptosis or alternative mechanisms) or cell senescence. However, in p53MUT cancer, p53 loses its WT function and in some cases may gain MUT function, either way this allows survival rather than growth inhibition, but the lack of cell cycle arrest also results in replication of DNA on a damaged template and hence further mutations. In some cases of p53WT cancer, WT p53 is suppressed by its negative regulators such as MDM2 and wild-type p53-induced phosphatase 1 (WIP1) (PPM1D). Either p53MUT or p53WT suppression by MDM2 is proposed to increase gemcitabine resistance (Figure 2) [55].
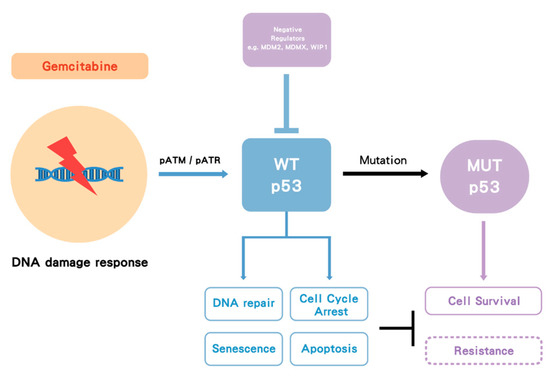
Figure 2. The proposed model of p53 impact on gemcitabine treatment. Gemcitabine induces a DNA damage response followed by activation of pATM/pATR and p53. Wild-type (WT) p53 activation can facilitate DNA repair, cell cycle arrest, apoptosis and senescence. Once p53 becomes mutated (MUT), it loses its WT function, and some mutant forms may gain function, which increases cell survival. In some cases, WT p53 is suppressed by its negative regulators such as MDM2/MDMX. Either p53 mutation or suppression by MDM2 may increase gemcitabine resistance. Dashed box indicates hypothesis.
However, studies regarding the role of p53 in gemcitabine resistance in BTCs are limited. As both pancreatic cancer and BTC have close anatomic location and similar histologic features, both were considered together in early studies. Palliative chemotherapy was demonstrated to improve both survival and quality of life for patients with advanced pancreatic cancer or BTC [56]. Gemcitabine became the standard treatment for both pancreatic cancer [57] and BTC [7]; therefore, studies related to gemcitabine resistance in pancreatic cancer may be applicable to BTCs and hence are discussed here.
Fiorini et al. demonstrated that p53MUT enhanced gemcitabine resistance in pancreatic cancer. Gemcitabine stabilized both nuclear p53WT and p53MUT and only the latter (p53MUT) induced chemoresistance and hyperproliferation evidenced by siRNA-mediated knockdown induced chemosensitivity and apoptosis in p53MUT but not in p53WT cell lines. This may be associated with overexpression of p53MUT-dependent Cdk1 and CCNB1 genes. However, a synergistic effect of the combination with gemcitabine and "p53-reactivating" compounds (CP-31398 and RITA) was found in both p53WT and p53MUT pancreatic cancer cell lines, which may have been due to a mechanism that did not involve p53 [58]. In a mouse model of pancreatic cancer, Wornann et al. reported that loss of p53WT function increased resistance to gemcitabine via activation of the JAK2–STAT3 pathway [59]. In a study by Dhyat et al. (2016) microRNA (miR) profiling in gemcitabine-resistant p53MUT pancreatic cancer cell lines suggested the involvement of miRs in pathways controlling cell cycle, proliferation, and apoptosis. In-silico analysis in this study indicated that some of the dysregulated miRs were regulated by p53MUT. In addition, MRP-1, Bcl-2 and CDK-1 were predicted to be targets of the dysregulated miRs and significant overexpression of MRP-1 and Bcl-2 was seen in the resistant cell clones [60]. These observations suggest that p53MUT may enhance chemoresistance via the dysregulation of miRs and that this might be associated with MRP-1 and Bcl-2 overexpression in p53MUT pancreatic cancer.
In the clinical setting, a phase III study (CONKO-001) evaluated the efficacy of adjuvant gemcitabine in resected pancreatic cancer. Next generation DNA sequencing was performed in this trial to identify clinically relevant prognostic and predictive mutations. In untreated patients, TP53 mutation was a negative prognostic factor for disease-free survival (DFS) (HR: 2.434, p = 0.005). Interestingly, however, TP53 mutation was a positive predictive factor for better DFS (HR: 0.235; p < 0.001 in p53MUT; HR: 0.794, p = 0.483 in p53WT) with gemcitabine treatment, with a significant test for interaction of the treatment status with TP53 mutational status (p = 0.003) [61]. Only subsets of patients with available samples were retrospectively analyzed, limiting the significance of this study, but the findings provide evidence for the role of p53MUT in pancreatic cancer, which may be relevant to BTC.
Further studies specific to BTC regarding p53 and chemoresistance are warranted to provide additional evidence, which would guide treatment. However, the above studies suggest a novel therapeutic strategy targeting both wild-type and mutant p53 tumors in combination with gemcitabine for pancreatic cancer as well as BTCs.
References
- Ustundag, Y.; Bayraktar, Y.; Cholangiocarcinoma: A compact review of the literature. World J. Gastroenterol. 2008, 14, 6458–6466.
- Khan, S.A. Cholangiocarcinoma. Lancet 2005, 366, 1303–1314.
- Patel, T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001, 33, 1353–1357.
- Wu, C.E.; Chen, M.H.; Yeh, C.N. mTOR Inhibitors in Advanced Biliary Tract Cancers. Int. J. Mol. Sci. 2019, 20, 500.
- Shaib, Y.; El-Serag, H.B. The epidemiology of cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 115–125.
- Wu, C.E. Prognostic and predictive factors for Taiwanese patients with advanced biliary tract cancer undergoing frontline chemotherapy with gemcitabine and cisplatin: A real-world experience. BMC Cancer 2020, 20, 422.
- Valle, J. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281.
- Hezel, A.F.; Deshpande, V.; Zhu, A.X. Genetics of Biliary Tract Cancers and Emerging Targeted Therapies. J. Clin. Oncol. 2010, 28, 3531–3540.
- Zhu, A.X.; Hezel, A.F. Development of Molecularly Targeted Therapies in Biliary Tract Cancers: Reassessing the Challenges and Opportunities. Hepatology 2011, 53, 695–704.
- Sahu, S.; Sun, W. Targeted therapy in biliary tract cancers-current limitations and potentials in the future. J Gastrointest. Oncol. 2017, 8, 324–336.
- Lee, J. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188.
- Malka, D. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828.
- Valle, J.W. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978.
- Chen, J.S. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann. Oncol. 2015, 26, 943–949.
- Leone, F. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-type KRAS advanced biliary tract cancer: A randomized phase 2 trial (Vecti-BIL study). Cancer 2016, 122, 574–581.
- Filippi, R. Pharmacotherapeutic options for biliary tract cancer: Current standard of care and new perspectives. Expert Opin. Pharmacother. 2019, 20, 2121–2137.
- Wu, C.E. Chemotherapy with gemcitabine plus cisplatin in patients with advanced biliary tract carcinoma at Chang Gung Memorial Hospital: A retrospective analysis. Chang. Gung Med. J. 2012, 35, 420–427.
- Farshidfar, F. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880.
- Mahipal, A. FGFR2 genomic aberrations: Achilles heel in the management of advanced cholangiocarcinoma. Cancer Treat. Rev. 2019, 78, 1–7.
- Abou-Alfa, G.K. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684.
- Piha-Paul, S.A.; Oh, D.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 2020, 147, 2190–2198.
- Goldstein, D.; Lemech, C.; Valle, J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017, 2 (Suppl. S1), e000152.
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413.
- Winkelmann, R.; Schneider, M.; Hartmann, S.; Schnitzbauer, A.A.; Zeuzem, S.; Peveling-Oberhag, J.; Hansmann, M.-L.; Walter, D. Microsatellite Instability Occurs Rarely in Patients with Cholangiocarcinoma: A Retrospective Study from a German Tertiary Care Hospital. Int. J. Mol. Sci. 2018, 19, 1421.
- Goeppert, B.; Roessler, S.; Renner, M.; Singer, S.; Mehrabi, A.; Vogel, M. N.; Pathil, A.; Czink, E.; Kohler, B.; Springfeld, C.; et al. Mismatch repair deficiency is a rare but putative therapeutically relevant finding in non-liver fluke associated cholangiocarcinoma. Br. J. Cancer 2019, 120, 109–114.
- Lane, D.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263.
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52.
- Kress, M.; et al. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J. Virol. 1979, 31, 472–483.
- Melero, J.A.; et al. Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology 1979, 93, 466–480.
- Smith, A.E.; Smith, R.; Paucha, E. Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 1979, 18, 335–346.
- Eliyahu, D.; et al. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 1984, 312, 646–649.
- Jenkins, J.R.; Rudge, K.; Currie, G.A. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 1984, 312, 651–654.
- Parada, L.F.; et al. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651.
- Eliyahu, D.; et al. Meth A fibrosarcoma cells express two transforming mutant p53 species. Oncogene 1988, 3, 313–321.
- Finlay, C.A.; et al. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol. Cell. Biol. 1988, 8, 531–539.
- Baker, S.J.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221.
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093.
- Malkin, D.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238.
- Fields, S.; Jang, S. Presence of a potent transcription activating sequence in the p53 protein. Science 1990, 249, 1046–1049.
- Bargonetti, J.; et al. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 1991, 65, 1083–1091.
- el-Deiry, W.S.; et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825.
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299.
- Momand, J.; et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245.
- Oliner, J.D.; et al. Oncoprotein Mdm2 Conceals the Activation Domain of Tumor Suppressor-P53. Nature 1993, 362, 857–860.
- Shvarts, A.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357.
- Linares, L.K.; et al. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc. Natl. Acad. Sci. USA 2003, 100, 12009–12014.
- Jones, S.N.; et al. Rescue of Embryonic Lethality in Mdm2-Deficient Mice by Absence of P53. Nature 1995, 378, 206–208.
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309.
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456.
- Brown, C.J.; et al. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873.
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008.
- Hollstein, M.; et al. p53 mutations in human cancers. Science 1991, 253, 49–53.
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96.
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8.
- Kocik, J., et al. Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers (Basel) 2019, 11, 1014.
- Glimelius, B.; et al. Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann. Oncol. 1996, 7, 593–600.
- Burris, H.A., 3rd; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413.
- Fiorini, C.; et al. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta 2015, 1853, 89–100.
- Wormann, S.M.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e12.
- Dhayat, S.A.; et al. MicroRNA Profiling Implies New Markers of Gemcitabine Chemoresistance in Mutant p53 Pancreatic Ductal Adenocarcinoma. PLoS ONE 2015, 10, e0143755.
- Sinn, M.; et al. TP53 Mutations Predict Sensitivity to Adjuvant Gemcitabine in Patients with Pancreatic Ductal Adenocarcinoma: Next-Generation Sequencing Results from the CONKO-001 Trial. Clin Cancer Res. 2020, 26, 3732–3739.

