 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tim Pawlik | -- | 2910 | 2022-10-12 07:44:40 | | | |
| 2 | Sirius Huang | Meta information modification | 2910 | 2022-10-12 08:12:26 | | |
Video Upload Options
Colorectal cancer remains one of the most significant sources of cancer-related morbidity and mortality worldwide. The liver is the most common site of metastatic spread. Multiple modalities exist to manage and potentially cure patients with metastatic colorectal cancer. Advances in genomic sequencing technology have greatly expanded our knowledge of colorectal cancer carcinogenesis and significantly reduced the cost and timing of the investigation.
1. Introduction
Colorectal cancer (CRC) is a leading cause of cancer-related morbidity and mortality worldwide [1]. While the global incidence continues to rise, disproportionately so in low- and middle-income countries, advances in CRC screening and multidisciplinary therapy have significantly improved mortality rates, especially among patients living in highly developed nations where mortality rates have declined in recent years [2]. Despite these improvements, CRC remains the second leading cause of cancer-related mortality in the United States [3].
Advances in molecular sequencing technology (e.g., next-generation sequencing (NGS)) and computational data analytics have greatly improved our understanding of CRC pathophysiology, revealing genomic variants responsible for CRC carcinogenesis. These molecular biomarkers, combined with the emerging fields of radiomics and artificial intelligence, provide clinicians with potentially actionable information. Current treatment algorithms for patients with CRLM incorporate certain biomarkers, including RAS, BRAF, and mismatch repair (MMR) status. Other biomarkers require validation on their clinical utility and are actively being investigated in clinical trials.
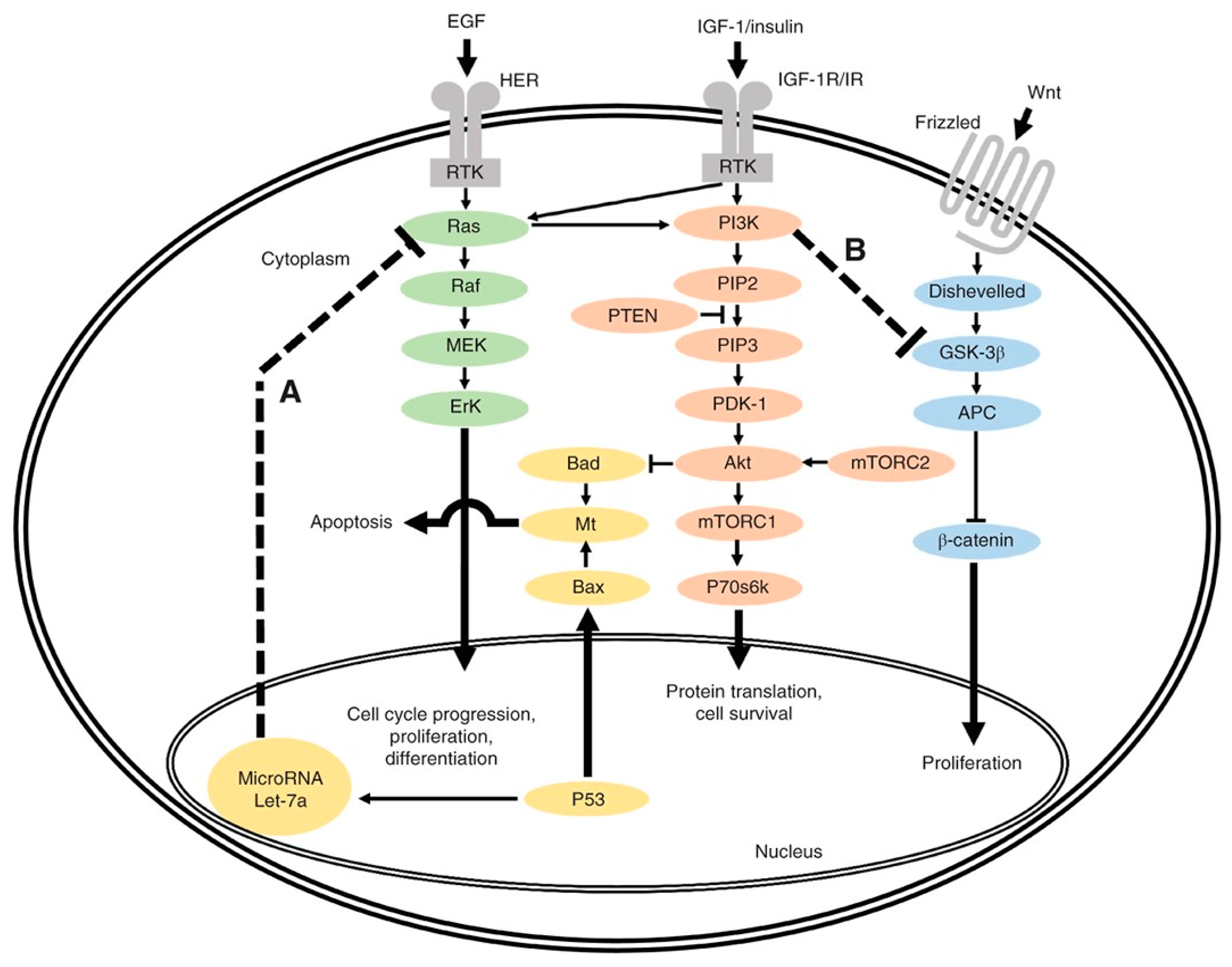
2. Genomic Biomarkers

3. Proteomic Biomarkers
4. Liquid Biopsy and ctDNA
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174.
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532.
- Del Vecchio, F.; Mastroiaco, V.; Di Marco, A.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next-generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246.
- Yamashita, S.; Chun, Y.S.; Kopetz, S.E.; Vauthey, J.N. Biomarkers in colorectal liver metastases. Br. J. Surg. 2018, 105, 618–627.
- Zhou, H.; Liu, Z.; Wang, Y.; Wen, X.; Amador, E.H.; Yuan, L.; Ran, X.; Xiong, L.; Ran, Y.; Chen, W.; et al. Colorectal liver metastasis: Molecular mechanism and interventional therapy. Signal Transduct. Target Ther. 2022, 7, 70.
- Black, D.L. Protein diversity from alternative splicing: A challenge for bioinformatics and post-genome biology. Cell 2000, 103, 367–370.
- Fahrner, M.; Bronsert, P.; Fichtner-Feigl, S.; Jud, A.; Schilling, O. Proteome biology of primary colorectal carcinoma and corresponding liver metastases. Neoplasia. 2021, 23, 1240–1251.
- Liu, X.; Xu, D.; Liu, Z.; Li, Y.; Zhang, C.; Gong, Y.; Jiang, Y.; Xing, B. THBS1 facilitates colorectal liver metastasis through enhancing epithelial-mesenchymal transition. Clin. Transl. Oncol. 2020, 22, 1730–1740.
- Kim, E.K.; Song, M.J.; Jung, Y.; Lee, W.S.; Jang, H.H. Proteomic Analysis of Primary Colon Cancer and Synchronous Solitary Liver Metastasis. Cancer Genom. Proteom. 2019, 16, 583–592.
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713.
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216.
- National Comprehensive Cancer Network. NCCN Guildeline: Colon Cancer (Version 1.2022); NCCN: Philadelphia, PA, USA, 2022.
- Testa, U.; Castelli, G.; Pelosi, E. Genetic Alterations of Metastatic Colorectal Cancer. Biomedicines 2020, 8, 414.
- Schirripa, M.; Bergamo, F.; Cremolini, C.; Casagrande, M.; Lonardi, S.; Aprile, G.; Yang, D.; Marmorino, F.; Pasquini, G.; Sensi, E.; et al. BRAF and RAS mutations as prognostic factors in metastatic colorectal cancer patients undergoing liver resection. Br. J. Cancer 2015, 112, 1921–1928.
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome. Biol. 2014, 15, 454.
- Vauthey, J.N.; Zimmitti, G.; Kopetz, S.E.; Shindoh, J.; Chen, S.S.; Andreou, A.; Curley, S.A.; Aloia, T.A.; Maru, D.M. RAS mutation status predicts survival and patterns of recurrence in patients undergoing hepatectomy for colorectal liver metastases. Ann. Surg. 2013, 258, 619–626; discussion 617–626.
- Connor, J.M.; Sanchez Loria, F.; Ardiles, V.; Grondona, J.; Sanchez, P.; Andriani, O.; Fauda, M.; Brancato, F.; Huertas, E.; Alvarez, F.; et al. Prognostic impact of K-RAS mutational status and primary tumor location in patients undergoing resection for colorectal cancer liver metastases: An update. Future Oncol. 2019, 15, 3149–3157.
- Siena, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Balfour, J.; Bardelli, A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 1308–1324.
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536.
- Serova, M.; Astorgues-Xerri, L.; Bieche, I.; Albert, S.; Vidaud, M.; Benhadji, K.A.; Emami, S.; Vidaud, D.; Hammel, P.; Theou-Anton, N.; et al. Epithelial-to-mesenchymal transition and oncogenic Ras expression in resistance to the protein kinase Cbeta inhibitor enzastaurin in colon cancer cells. Mol. Cancer Ther. 2010, 9, 1308–1317.
- Odisio, B.C.; Yamashita, S.; Huang, S.Y.; Harmoush, S.; Kopetz, S.E.; Ahrar, K.; Shin Chun, Y.; Conrad, C.; Aloia, T.A.; Gupta, S.; et al. Local tumour progression after percutaneous ablation of colorectal liver metastases according to RAS mutation status. Br. J. Surg. 2017, 104, 760–768.
- Zhang, Q.; Peng, J.; Ye, M.; Weng, W.; Tan, C.; Ni, S.; Huang, D.; Sheng, W.; Wang, L. KRAS Mutation Predicted More Mirometastases and Closer Resection Margins in Patients with Colorectal Cancer Liver Metastases. Ann. Surg. Oncol. 2020, 27, 1164–1173.
- Brudvik, K.W.; Mise, Y.; Chung, M.H.; Chun, Y.S.; Kopetz, S.E.; Passot, G.; Conrad, C.; Maru, D.M.; Aloia, T.A.; Vauthey, J.N. RAS Mutation Predicts Positive Resection Margins and Narrower Resection Margins in Patients Undergoing Resection of Colorectal Liver Metastases. Ann. Surg. Oncol. 2016, 23, 2635–2643.
- Margonis, G.A.; Sergentanis, T.N.; Ntanasis-Stathopoulos, I.; Andreatos, N.; Tzanninis, I.G.; Sasaki, K.; Psaltopoulou, T.; Wang, J.; Buettner, S.; Papalois, A.; et al. Impact of Surgical Margin Width on Recurrence and Overall Survival Following R0 Hepatic Resection of Colorectal Metastases: A Systematic Review and Meta-analysis. Ann. Surg. 2018, 267, 1047–1055.
- Margonis, G.A.; Sasaki, K.; Andreatos, N.; Kim, Y.; Merath, K.; Wagner, D.; Wilson, A.; Buettner, S.; Amini, N.; Antoniou, E.; et al. KRAS Mutation Status Dictates Optimal Surgical Margin Width in Patients Undergoing Resection of Colorectal Liver Metastases. Ann. Surg. Oncol. 2017, 24, 264–271.
- Hatta, A.A.Z.; Pathanki, A.M.; Hodson, J.; Sutcliffe, R.P.; Marudanayagam, R.; Roberts, K.J.; Chatzizacharias, N.; Isaac, J.; Muiesan, P.; Taniere, P.; et al. The effects of resection margin and KRAS status on outcomes after resection of colorectal liver metastases. HPB 2021, 23, 90–98.
- Bertsimas, D.; Margonis, G.A.; Sujichantararat, S.; Boerner, T.; Ma, Y.; Wang, J.; Kamphues, C.; Sasaki, K.; Tang, S.; Gagniere, J.; et al. Using Artificial Intelligence to Find the Optimal Margin Width in Hepatectomy for Colorectal Cancer Liver Metastases. JAMA Surg. 2022, 157, e221819.
- Pikouli, A.; Papaconstantinou, D.; Wang, J.; Kavezou, F.; Pararas, N.; Nastos, C.; Pikoulis, E.; Margonis, G.A. Reevaluating the prognostic role of BRAF mutation in colorectal cancer liver metastases. Am. J. Surg. 2022, 223, 879–883.
- Margonis, G.A.; Buettner, S.; Andreatos, N.; Kim, Y.; Wagner, D.; Sasaki, K.; Beer, A.; Schwarz, C.; Løes, I.M.; Smolle, M.; et al. Association of BRAF Mutations With Survival and Recurrence in Surgically Treated Patients With Metastatic Colorectal Liver Cancer. JAMA Surg. 2018, 153, e180996.
- Gagnière, J.; Dupré, A.; Gholami, S.S.; Pezet, D.; Boerner, T.; Gönen, M.; Kingham, T.P.; Allen, P.J.; Balachandran, V.P.; De Matteo, R.P.; et al. Is Hepatectomy Justified for BRAF Mutant Colorectal Liver Metastases?: A Multi-institutional Analysis of 1497 Patients. Ann. Surg. 2020, 271, 147–154.
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594.
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630.
- Ross, J.S.; Fakih, M.; Ali, S.M.; Elvin, J.A.; Schrock, A.B.B.; Suh, J.; Vergilio, J.A.; Ramkissoon, S.; Severson, E.; Daniel, S.; et al. Targeting HER2 in colorectal cancer: The landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer 2018, 124, 1358–1373.
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet. Oncol. 2019, 20, 518–530.
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet. Oncol. 2016, 17, 738–746.
- Gelsomino, F.; Barbolini, M.; Spallanzani, A.; Pugliese, G.; Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat. Rev. 2016, 51, 19–26.
- Cohen, R.; Buhard, O.; Cervera, P.; Hain, E.; Dumont, S.; Bardier, A.; Bachet, J.B.; Gornet, J.M.; Lopez-Trabada, D.; Kaci, R.; et al. Clinical and molecular characterisation of hereditary and sporadic metastatic colorectal cancers harbouring microsatellite instability/DNA mismatch repair deficiency. Eur. J. Cancer 2017, 86, 266–274.
- Kim, C.G.; Ahn, J.B.; Jung, M.; Beom, S.H.; Kim, C.; Kim, J.H.; Heo, S.J.; Park, H.S.; Kim, N.K.; Min, B.S.; et al. Effects of microsatellite instability on recurrence patterns and outcomes in colorectal cancers. Br. J. Cancer 2016, 115, 25–33.
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218.
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103.
- Chun, Y.S.; Passot, G.; Yamashita, S.; Nusrat, M.; Katsonis, P.; Loree, J.M.; Conrad, C.; Tzeng, C.D.; Xiao, L.; Aloia, T.A.; et al. Deleterious Effect of RAS and Evolutionary High-risk TP53 Double Mutation in Colorectal Liver Metastases. Ann. Surg. 2019, 269, 917–923.
- Yamashita, S.; Chun, Y.S.; Kopetz, S.E.; Maru, D.; Conrad, C.; Aloia, T.A.; Vauthey, J.N. APC and PIK3CA Mutational Cooperativity Predicts Pathologic Response and Survival in Patients Undergoing Resection for Colorectal Liver Metastases. Ann. Surg. 2020, 272, 1080–1085.
- Deming, D.A.; Leystra, A.A.; Nettekoven, L.; Sievers, C.; Miller, D.; Middlebrooks, M.; Clipson, L.; Albrecht, D.; Bacher, J.; Washington, M.K.; et al. PIK3CA and APC mutations are synergistic in the development of intestinal cancers. Oncogene 2014, 33, 2245–2254.
- Wong, G.Y.M.; Diakos, C.; Hugh, T.J.; Molloy, M.P. Proteomic Profiling and Biomarker Discovery in Colorectal Liver Metastases. Int. J. Mol. Sci. 2022, 23, 6091.
- Ku, X.; Xu, Y.; Cai, C.; Yang, Y.; Cui, L.; Yan, W. In-Depth Characterization of Mass Spectrometry-Based Proteomic Profiles Revealed Novel Signature Proteins Associated with Liver Metastatic Colorectal Cancers. Anal. Cell Pathol. 2019, 2019, 7653230.
- Li, Q.; Li, Y.; Xu, J.; Wang, S.; Xu, Y.; Li, X.; Cai, S. Aldolase B Overexpression is Associated with Poor Prognosis and Promotes Tumor Progression by Epithelial-Mesenchymal Transition in Colorectal Adenocarcinoma. Cell Physiol. Biochem. 2017, 42, 397–406.
- Yang, Q.; Bavi, P.; Wang, J.Y.; Roehrl, M.H. Immuno-proteomic discovery of tumor tissue autoantigens identifies olfactomedin 4, CD11b, and integrin alpha-2 as markers of colorectal cancer with liver metastases. J. Proteomics 2017, 168, 53–65.
- Kwon, M.; Lee, S.J.; Wang, Y.; Rybak, Y.; Luna, A.; Reddy, S.; Adem, A.; Beaty, B.T.; Condeelis, J.S.; Libutti, S.K. Filamin A interacting protein 1-like inhibits WNT signaling and MMP expression to suppress cancer cell invasion and metastasis. Int J. Cancer 2014, 135, 48–60.
- Seetoo, D.Q.; Crowe, P.J.; Russell, P.J.; Yang, J.L. Quantitative expression of protein markers of plasminogen activation system in prognosis of colorectal cancer. J. Surg. Oncol. 2003, 82, 184–193.
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200.
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406.
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518.
- Rao, V.S.; Gu, Q.; Tzschentke, S.; Lin, K.; Ganig, N.; Thepkaysone, M.L.; Wong, F.C.; Polster, H.; Seifert, L.; Seifert, A.M.; et al. Extravesicular TIMP-1 is a non-invasive independent prognostic marker and potential therapeutic target in colorectal liver metastases. Oncogene 2022, 41, 1809–1820.
- Shen, Z.; Wang, B.; Luo, J.; Jiang, K.; Zhang, H.; Mustonen, H.; Puolakkainen, P.; Zhu, J.; Ye, Y.; Wang, S. Global-scale profiling of differential expressed lysine acetylated proteins in colorectal cancer tumors and paired liver metastases. J. Proteom. 2016, 142, 24–32.
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783.
- Pettini, F.; Visibelli, A.; Cicaloni, V.; Iovinelli, D.; Spiga, O. Multi-Omics Model Applied to Cancer Genetics. Int. J. Mol. Sci. 2021, 22, 5751.
- Li, C.; Sun, Y.D.; Yu, G.Y.; Cui, J.R.; Lou, Z.; Zhang, H.; Huang, Y.; Bai, C.G.; Deng, L.L.; Liu, P.; et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell 2020, 38, 734–747.e739.
- Ma, Y.S.; Huang, T.; Zhong, X.M.; Zhang, H.W.; Cong, X.L.; Xu, H.; Lu, G.X.; Yu, F.; Xue, S.B.; Lv, Z.W.; et al. Proteogenomic characterization and comprehensive integrative genomic analysis of human colorectal cancer liver metastasis. Mol. Cancer 2018, 17, 139.
- Ma, Y.S.; Wu, Z.J.; Zhang, H.W.; Cai, B.; Huang, T.; Long, H.D.; Xu, H.; Zhao, Y.Z.; Yin, Y.Z.; Xue, S.B.; et al. Dual Regulatory Mechanisms of Expression and Mutation Involving Metabolism-Related Genes FDFT1 and UQCR5 during CLM. Mol. Ther. Oncolytics 2019, 14, 172–178.
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421.
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153.
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548.
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586.
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224.
- Barimani, D.; Kauppila, J.H.; Sturesson, C.; Sparrelid, E. Imaging in disappearing colorectal liver metastases and their accuracy: A systematic review. World J. Surg. Oncol. 2020, 18, 264.
- Murahashi, S.; Akiyoshi, T.; Sano, T.; Fukunaga, Y.; Noda, T.; Ueno, M.; Zembutsu, H. Serial circulating tumour DNA analysis for locally advanced rectal cancer treated with preoperative therapy: Prediction of pathological response and postoperative recurrence. Br. J. Cancer 2020, 123, 803–810.
- Zhou, J.; Wang, C.; Lin, G.; Xiao, Y.; Jia, W.; Xiao, G.; Liu, Q.; Wu, B.; Wu, A.; Qiu, H.; et al. Serial Circulating Tumor DNA in Predicting and Monitoring the Effect of Neoadjuvant Chemoradiotherapy in Patients with Rectal Cancer: A Prospective Multicenter Study. Clin. Cancer Res. 2021, 27, 301–310.
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2019, 68, 663–671.
- Vidal, J.; Casadevall, D.; Bellosillo, B.; Pericay, C.; Garcia-Carbonero, R.; Losa, F.; Layos, L.; Alonso, V.; Capdevila, J.; Gallego, J.; et al. Clinical Impact of Presurgery Circulating Tumor DNA after Total Neoadjuvant Treatment in Locally Advanced Rectal Cancer: A Biomarker Study from the GEMCAD 1402 Trial. Clin. Cancer Res. 2021, 27, 2890–2898.
- Khakoo, S.; Carter, P.D.; Brown, G.; Valeri, N.; Picchia, S.; Bali, M.A.; Shaikh, R.; Jones, T.; Begum, R.; Rana, I.; et al. MRI Tumor Regression Grade and Circulating Tumor DNA as Complementary Tools to Assess Response and Guide Therapy Adaptation in Rectal Cancer. Clin. Cancer Res. 2020, 26, 183–192.
- Mason, M.C.; Tzeng, C.D.; Tran Cao, H.S.; Aloia, T.A.; Newhook, T.E.; Overman, M.J.; Kopetz, S.E.; Vauthey, J.N.; Chun, Y.S. Preliminary Analysis of Liquid Biopsy after Hepatectomy for Colorectal Liver Metastases. J. Am. Coll. Surg. 2021, 233, 82–89.e81.
- Tie, J.; Wang, Y.; Cohen, J.; Li, L.; Hong, W.; Christie, M.; Wong, H.L.; Kosmider, S.; Wong, R.; Thomson, B.; et al. Circulating tumor DNA dynamics and recurrence risk in patients undergoing curative intent resection of colorectal cancer liver metastases: A prospective cohort study. PLoS. Med. 2021, 18, e1003620.
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801.
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435.
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Fenocchio, E.; Amatu, A.; Corallo, S.; Manai, C.; Tosi, F.; et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: The CHRONOS trial. J. Clin. Oncol. 2021, 39, 3506.
- Martinelli, E.; Martini, G.; Famiglietti, V.; Troiani, T.; Napolitano, S.; Pietrantonio, F.; Ciardiello, D.; Terminiello, M.; Borrelli, C.; Vitiello, P.P.; et al. Cetuximab Rechallenge Plus Avelumab in Pretreated Patients With RAS Wild-type Metastatic Colorectal Cancer: The Phase 2 Single-Arm Clinical CAVE Trial. JAMA Oncol. 2021, 7, 1529–1535.
- Herberts, C.; Wyatt, A.W. Technical and biological constraints on ctDNA-based genotyping. Trends Cancer 2021, 7, 995–1009.


