Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Deborah Marsh | -- | 2252 | 2022-10-12 02:26:15 | | | |
| 2 | Camila Xu | Meta information modification | 2252 | 2022-10-12 02:54:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Xie, T.; Dickson, K.; Yee, C.; Ma, Y.; Ford, C.E.; Bowden, N.A.; Marsh, D.J. Poly ADP Ribose Polymerase Inhibitors. Encyclopedia. Available online: https://encyclopedia.pub/entry/28951 (accessed on 25 June 2026).
Xie T, Dickson K, Yee C, Ma Y, Ford CE, Bowden NA, et al. Poly ADP Ribose Polymerase Inhibitors. Encyclopedia. Available at: https://encyclopedia.pub/entry/28951. Accessed June 25, 2026.
Xie, Tao, Kristie-Ann Dickson, Christine Yee, Yue Ma, Caroline E. Ford, Nikola A. Bowden, Deborah J. Marsh. "Poly ADP Ribose Polymerase Inhibitors" Encyclopedia, https://encyclopedia.pub/entry/28951 (accessed June 25, 2026).
Xie, T., Dickson, K., Yee, C., Ma, Y., Ford, C.E., Bowden, N.A., & Marsh, D.J. (2022, October 12). Poly ADP Ribose Polymerase Inhibitors. In Encyclopedia. https://encyclopedia.pub/entry/28951
Xie, Tao, et al. "Poly ADP Ribose Polymerase Inhibitors." Encyclopedia. Web. 12 October, 2022.
Copy Citation
Poly ADP ribose polymerases (PARPs) are a family of enzymes that catalyse the transfer of ADP-ribose to target proteins, functioning in fundamental cellular processes including transcription, chromatin remodelling and DNA repair. PARP inhibitors (PARPis) including olaparib, niraparib and rucaparib are approved for the clinical management of women with ovarian cancer.
ovarian cancer
high-grade serous ovarian cancer
PARP
homologous recombination repair
BRCA
homologous recombination deficiency
synthetic lethal
olaparib
niraparib
rucaparib
1. Introduction
The application of molecular targeted therapy to treat poor outcome malignancies is revolutionising the field of cancer medicine and extending the lives of patients. Fundamental discovery science has elucidated the cellular response to DNA damage and this knowledge has been harnessed for the development of new therapies. Acknowledged as a major breakthrough, blocking poly (ADP-ribose) polymerase (PARP), a key enzyme in DNA repair, in tumours with genetic or epigenetic abrogation of proteins involved in homologous recombination repair (HRR), creates a synthetic lethal phenotype that kills cancer cells. Small molecule inhibitors of PARP are now clinically approved in many countries for the treatment of a number of malignancies including breast, ovarian and pancreatic cancers. In fact, in a relatively short time, PARP inhibitors (PARPis) have entirely altered the approach to treating a large subset of ovarian cancers. The clinical use of PARPis represents a major and impactful advance in the management of this disease.
1.1. Ovarian Cancer and Defects in Homologous Recombination Repair (HRR)
Ovarian cancer encompasses a number of distinct malignancies that share an anatomical location, yet have different cellular origins, molecular profiles and responses to therapy [1][2][3][4][5]. High-grade serous ovarian cancer (HGSOC) is the most common histological subtype of epithelial ovarian cancer, with less common subtypes including ovarian clear cell carcinoma (OCCC), endometrioid ovarian cancer (EnOC), mucinous ovarian cancer (MOC) and low-grade serous ovarian cancer (LGSOC). An additional exceedingly rare subtype is small cell carcinoma of the ovary, hypercalcaemic type (SCCOHT). This research focuses on HGSOC, an aggressive malignancy, generally treated with a combination of surgery and platinum-taxane based chemotherapy. Despite this treatment regime, the majority of women relapse within two years and recurrent disease is generally viewed as incurable [6]. Five-year survival has remained less than 50% and, until very recently, options for treatment in addition to chemotherapy have been absent [7].
Molecular profiling is revolutionising the clinical management of HGSOC, with actionable targets now known. Mutation of the tumour suppressor gene TP53 occurs in almost 100% of HGSOC [8][9]. Extensive research efforts are ongoing worldwide to target mutant p53 in ovarian and many other malignancies, for example using compounds that reactivate mutant p53 back to its wild-type form such as APR-246 (also known as PRIMA-1MET) [10][11][12]. To date, no mutant p53 targeting drug has been approved for routine clinical use. In sharp contrast to this are the recent successes of clinical targeting of molecular defects in the HRR pathway. Over 50% of HGSOC have a defect in this pathway due to mutations in BRCA1, BRCA2, RAD51C, RAD51D, ATM or PALB2 [8][13][14][15][16][17], or methylation of genes including BRCA1 [8][18][19][20][21][22] or RAD51C [23][24][25]. Mutations in HRR-associated genes have also been identified in OCCC and EnOC, albeit at lower frequencies than for HGSOC [26][27][28].
Collectively, tumours with a deficient HRR pathway due to genetic or epigenetic events are described as having a “BRCAness” phenotype that is frequently accompanied by higher levels of loss of heterozygosity, telomeric allelic imbalance and large-scale state transitions, due to the cell’s impaired ability to repair double strand breaks (DSBs), referred to as a genomic scar [29][30][31]. This genomic instability present as the result of BRCAness can be measured and used as a diagnostic tool for identifying HRR deficiency in tumours. By analysing these phenotypic effects of HRR deficiency, the involvement of defective HRR genes other than BRCA1 or BRCA2 can also be identified by implication. This includes HRR genes where their expression is determined by methylation. Commercial FDA-approved companion diagnostic (CDx) tests, FoundationOne® CDx (Foundation Medicine, Cambridge, MA, USA) and myChoice® CDx (Myriad, Salt Lake City, UT, USA) are now used to determine whether a woman with ovarian cancer is likely to see clinical benefit from a PARPi based on having HRR deficiency [32][33]. These tests generate a score, above which the tumour is likely HRR deficient and below which HRR proficient.
In addition to certain ovarian cancers, BRCAness is seen in other malignancies including breast [34], prostate [35], pancreatic [36], gastric [37] and colorectal cancers [38], as well as in acute leukemias [39]. While BRCAness is a clear driver of malignancy, it can be targeted using synthetic lethal strategies that involve inhibition of PARP.
1.2. The PARP Family and DNA Repair
The 17-member PARP family of enzymes includes PARP1, PARP2, PARP3, PARP4 (also known as Vault PARP) and tankyrases 1 and 2 (PARP5a and PARP5b) [40][41][42]. PARPs are involved in many key cellular processes including regulating transcription, translation, telomere maintenance, remodelling the chromatin landscape and, importantly in the context of this research, DNA repair [43][44]. PARPs catalyse the transfer of poly (ADP-ribose) (poly(adenosinediphosphate-ribose)) to target proteins. In this process of polyADP-ribosylation, also known as PARylation, catalytic activation of PARP synthesises poly (ADP-ribose), PAR, from its substrate nicotinamide adenine dinucleotide (NAD+) to form chains of PAR polymers. These chains attach covalently to specific amino acid residues on either PARP itself, known as auto-PARylation, or other acceptor proteins [45][46]. It is understood that PARP1 is responsible for over 90% of PARylation in the context of DNA damage, with PARPs 2, 3, 4, 5a and 5b also having PARylation activity [45][47].
During the DNA Damage Response (DDR) PARPs bind to sites of single strand breaks (SSBs) undergoing base excision repair (BER), PARylating substrates in order to facilitate recruitment of DNA repair machinery [48]. PARylation also destabilises PARP1 interaction with the SSB, uncoupling these two factors that then facilitates access for BER machinery [49]. Left unrepaired, SSBs pose a risk to genetic stability and therefore to cell survival. When the DNA replication fork encounters a SSB it can stall and collapse, causing a double strand break (DSB) that requires correction via HRR [50]. This creates a pharmacological opportunity in HRR deficient cells whereby inhibition of PARP reduces the ability of cells to repair DNA damage via the BER pathway. In this case, HRR deficient cells are unable to repair DNA damage by either HRR or BER, creating a synthetic lethal phenotype resulting in cancer cell death [51][52][53] (Figure 1). Simply stated, the combination of HRR deficiency and PARP inhibition is fatal to the cell. Further, the scaffold protein XRCC1 assembles protein complexes containing DNA polymerase β and DNA ligase III, preventing PARP1 engagement and activity during BER. This flags XRCC1 as an “anti-trapper” that may have implications for genome integrity [54].
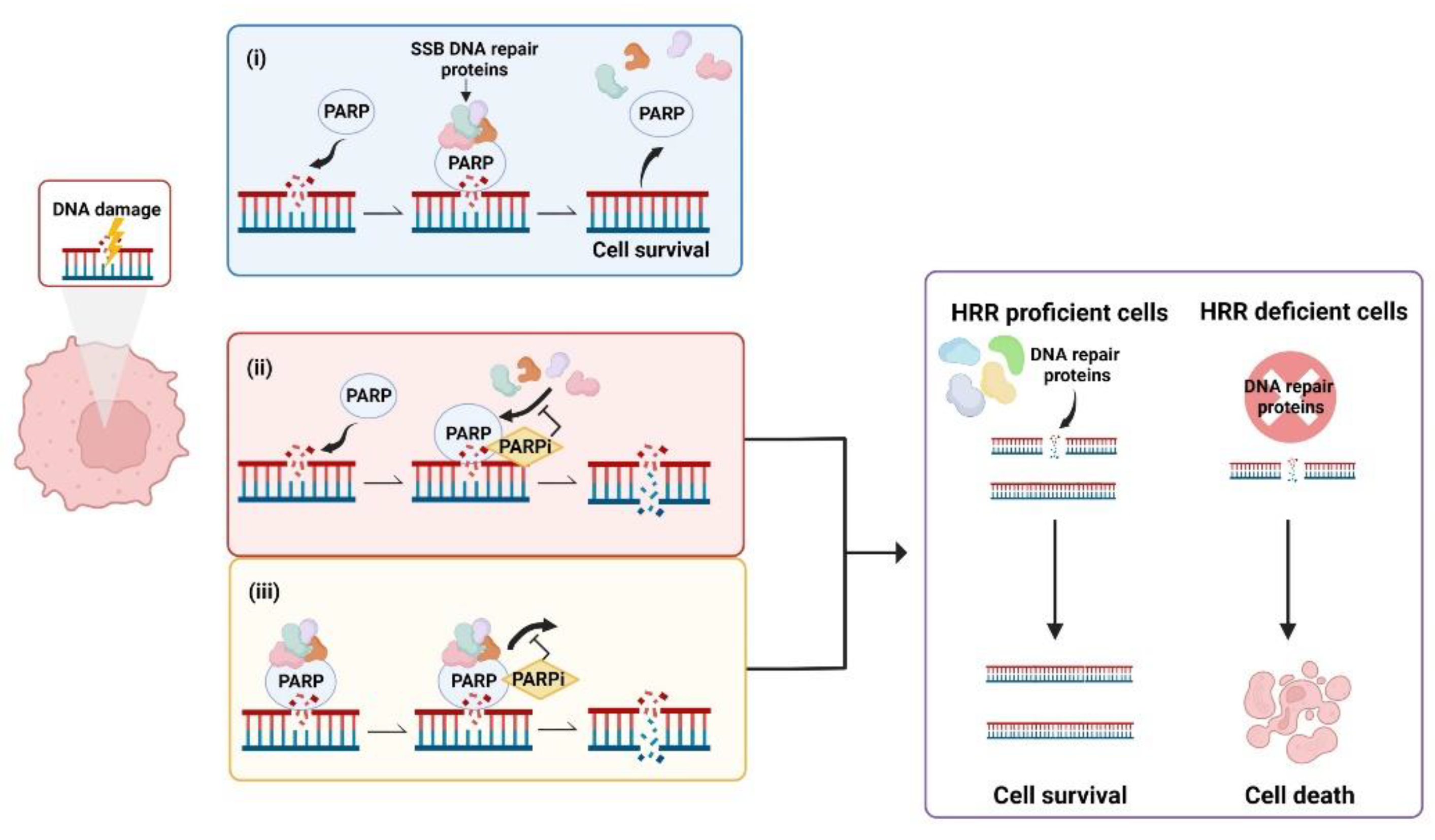
Figure 1. Synthetic lethality occurs when a defect in the homologous recombination repair (HRR) pathway is combined with inhibition of poly (ADP-ribose) polymerase (PARP). (i) PARP binds to sites of single strand breaks (SSBs), PARylates substrates and recruits DNA repair proteins. (ii) PARP inhibitors (PARPis) bind PARP, preventing PARylation and blocking access of PARP to DNA lesions that results in double strand breaks (DSBs). (iii) PARPis can also work to trap PARP at the DNA, inhibiting the dissociation of PARP from DNA and leading to the generation of DSBs. In cells with defective HRR, DSBs are unable to be repaired, leading to cell death. Created with Biorender.com.
2. PARP Inhibitors (PARPis)—Focus on Ovarian Cancer
PARP inhibitors (PARPis) including olaparib (Lynparza®; AstraZeneca Pharmaceuticals, Cambridge, UK), rucaparib (Rubraca®; Clovis Oncology, Inc., Boulder, CO, USA) and niraparib (Zejula®; GlaxoSmithKline, Brentford, Middlesex, UK) are small molecule inhibitors of PARP that have been approved by the US Food and Drug Administration (FDA), and other regulatory authorities worldwide, for women with ovarian cancer under certain conditions, including as maintenance therapy. Talazoparib (Talzenna®; Pfizer, Inc., Manhattan, NY, USA) is approved for treatment of advanced breast cancer and veliparib (ABT-888; AbbVie, North Chicago, IL, USA) is still being evaluated. An additional two PARPis, pamiparib (Partruvix™; BeiGene Ltd., Beijing, China) and fuzuloparib (AiRuiYi®, formerly fluzoparib; Jiangsu Hengrui Pharmaceuticals Co., Ltd., Lianyungang, China), have been approved in China for the treatment of women with ovarian cancer.
2.1. Timeline of Discovery and Clinical Adoption of PARPis
In 1963, Chambon and colleagues reported their initial discovery that nicotinamide mononucleotide enhanced the activity of a DNA dependent enzyme [55][56]. This discovery would go on to form the basis of the PARP field as we know it today. By the early 1980s, PARP had been found to play an essential role in the repair of DNA SSBs and the first PARPi was identified [57][58][59]. In 2005, the landmark discovery that BRCA dysfunction greatly sensitised cancer cells to PARP inhibition was reported [60][61]. This was proof that the concept of synthetic lethality could be adopted as a therapeutic strategy by targeting BRCA-related HRR dysfunction with a DNA repair inhibitor. For malignancies such as HGSOC where BRCA1 and BRCA2 mutations are prevalent [14], this discovery marked a clear turning point and new hope for molecular targeted therapy.
Almost a decade later in December 2014, olaparib became the first PARPi approved by the FDA for the treatment of advanced, recurrent ovarian cancers with germline BRCA mutation, or suspected germline mutation, and previous treatment of three or more lines of chemotherapy [62][63]. FDA approval of rucaparib followed in 2016, for treatment of the same indication [51][64]. Niraparib was approved by the FDA in 2017 for maintenance treatment of patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who were in complete or partial response to platinum-based chemotherapy [65]. Olaparib in 2017 [66] and rucaparib in 2018 [67] were also FDA approved as maintenance therapies under the same conditions as niraparib. While not currently approved for the treatment of ovarian cancer, in 2018 talazoparib was approved for the treatment of locally advanced or metastatic BRCA-mutated HER2-negative breast cancers [68]. Pamiparib was approved in China in 2021 for the treatment of germline BRCA mutated recurrent advanced ovarian, fallopian tube and primary peritoneal cancer in women who have had two or more lines of chemotherapy [69], as was fuzuloparib [70].
Reflecting the growing understanding that HRR-deficiency was the result of more than just BRCA1 or BRCA2 defects, niraparib was FDA approved for HRR-deficient advanced ovarian cancer in 2019 [71]. The combination of olaparib and the anti-angiogenic bevacizumab was FDA approved in 2020 for first-line maintenance of HRR deficient advanced epithelial ovarian, fallopian tube, or primary peritoneal cancers in complete or partial response to platinum-based chemotherapy [72]. Of significance, in 2020 the FDA-approved front-line maintenance with niraparib for platinum sensitive advanced ovarian cancer regardless of HRR status [73][74]. Other PARPis, including veliparib, are currently undergoing preclinical and clinical research and may be approved for either first-line or maintenance treatment of ovarian cancers in the future. A timeline of PARP and PARPi discovery, as well as clinical approvals is shown in Figure 2.
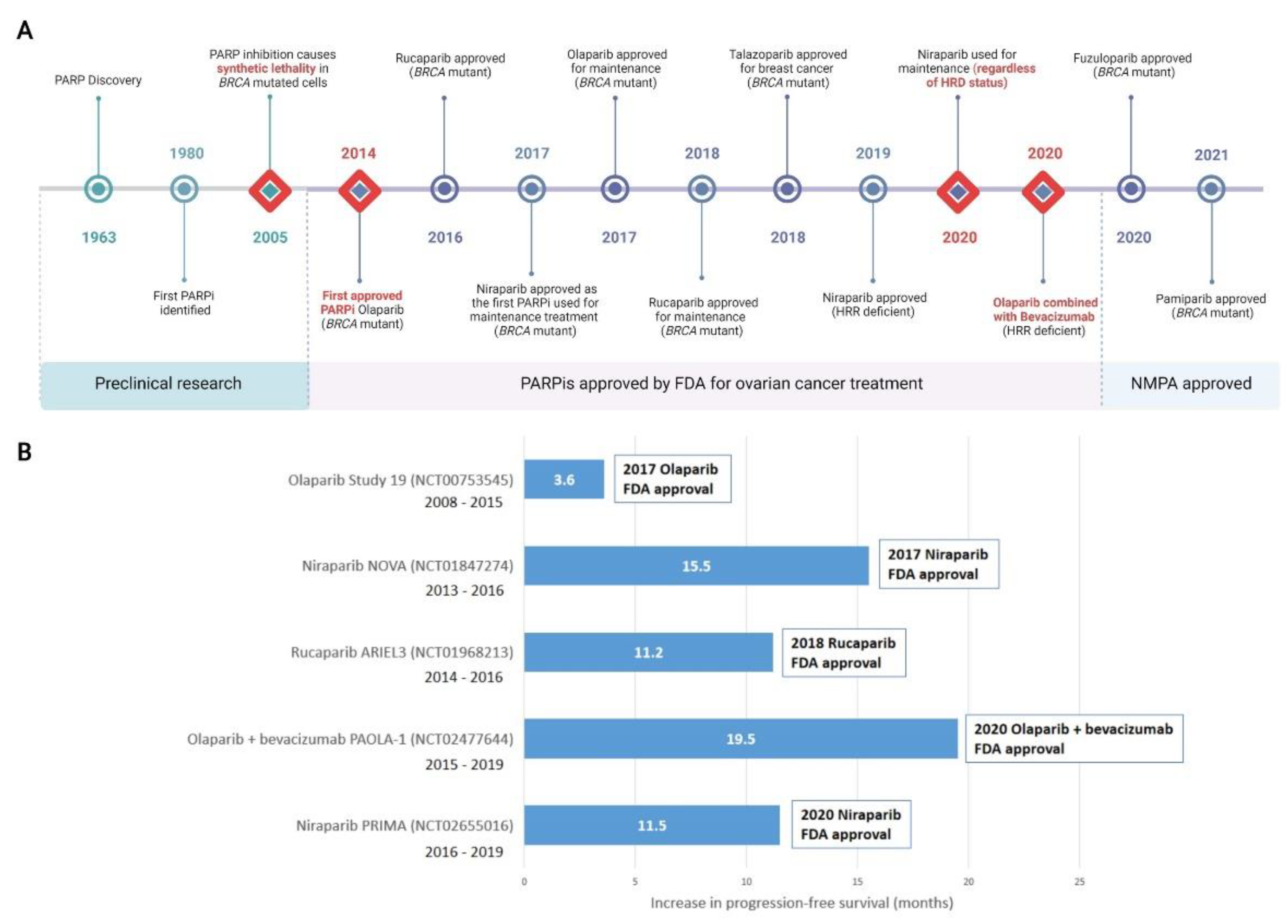
Figure 2. Discovery of PARP inhibitors and their introduction to the clinic. (A) Milestones in the discovery of PARP and clinical adoption of PARPis are recorded over time. Major milestones are indicated with a red diamond. All milestones are specific to the treatment of ovarian cancer, with the exception of talazoparib which was first approved for metastatic breast cancer and not currently approved for ovarian cancer. (B) Selected landmark clinical trials investigating progression-free survival (PFS) that were instrumental in clinical approval of PARPis, including in combination therapy, are shown. Increased PFS in treatment compared to placebo arms are represented. Additional months of PFS for treatment versus placebo groups are reported for the whole cohort (NCT00753545); BRCA mutant patients (NCT01847274 and NCT01968213); HRD positive patients including BRCA mutant (NCT02477644); HRD positive patients (NCT02655016). Detailed clinical trial information can be found in original content. For trials noted as active but not recruiting, the primary completion date for data collection is noted. FDA, Food and Drug Administration (US); NMPA, National Medical Products Administration (China). HRD, homologous recombination deficiency; HRR, Homologous Recombination Repair; PARPi, PARP inhibitor. Created with Biorender.com.
2.2. Structure and Function of PARPis
While all PARPis contain pharmacologically active nicotinamide/benzamide core structures that compete with endogenous NAD to access catalytic binding pockets of PARPs, each has a unique structure overall [75][76][77]. PARP1 and PARP2 are common targets for all PARPis; however, PARPis have different binding affinities for certain other PARP family members [78][79] (Figure 3). Antolin and colleagues summarise the affinity of olaparib, rucaparib, niraparib and talazoparib for different PARPs based on IC50 values from the literature and the ChEMBL database (www.ebi.ac.uk/chembl) [78]. PARP trapping potency also differs between PARPis. During PARP trapping, the PARP complex locks on at sites of DNA breakage, inhibiting the release of PARP and likely removing it from the process of DNA repair-associated PARylation, as well as inhibiting binding of DNA repair factors [80][81][82]. In order from highest to lowest, the PARP trapping abilities of five PARPis have been reported as talazoparib, niraparib, rucaparib, olaparib, and finally veliparib [80][81]. Pamiparib has also been reported to display PARP trapping activity [83]. It has been suggested that differences in PARP trapping activity associated with higher cytotoxicity will need to be considered when testing in combination with other cytotoxic therapies [80].
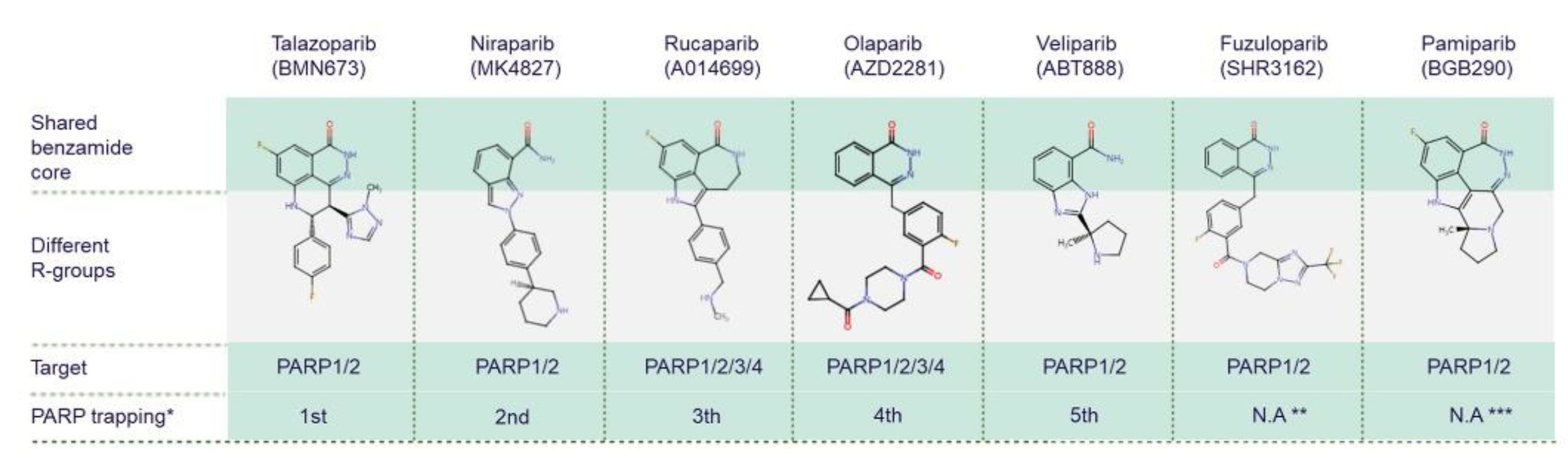
Figure 3. Chemical structures of clinically used PARP inhibitors. The chemical structures of PARP inhibitors (PARPis) are from ChEMBL database (www.ebi.ac.uk/chembl, accessed on 15 June 2022). * PARPis are listed by PARP trapping potency from highest to lowest [80]. ** There is no currently available data (N.A) for fuzuloparib PARP trapping potency. *** There is no currently available data for pamiparib trapping potency relative to other PARPis.
In addition to roles in DNA repair, PARPs function in other critical cellular processes. A role for PARP1 and PARP2 has been described in the maintenance of T-lymphocyte number and function [84]. PARP1 trapping has been shown to result in toxicity in healthy bone marrow [85]. PARP2 has been implicated in erythropoiesis and PARP2 deficient mice (Parp2−/−) are chronically anaemic [86]. Given these additional roles of PARP family members in important cellular processes, it is perhaps not surprising that adverse events are reported by patients taking PARPis.
References
- Yee, C.; Dickson, K.A.; Muntasir, M.N.; Ma, Y.; Marsh, D.J. Three-Dimensional Modelling of Ovarian Cancer: From Cell Lines to Organoids for Discovery and Personalized Medicine. Front. Bioeng. Biotechnol. 2022, 10, 836984.
- Dion, L.; Carton, I.; Jaillard, S.; Nyangoh Timoh, K.; Henno, S.; Sardain, H.; Foucher, F.; Levêque, J.; de la Motte Rouge, T.; Brousse, S.; et al. The Landscape and Therapeutic Implications of Molecular Profiles in Epithelial Ovarian Cancer. J. Clin. Med. 2020, 9, 2239.
- Kurman, R.J.; Shih Ie, M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443.
- Köbel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Ionescu, D.N.; Rajput, A.; et al. Ovarian carcinoma subtypes are different diseases: Implications for biomarker studies. PLoS Med. 2008, 5, e232.
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679.
- Beesley, V.L.; Green, A.C.; Wyld, D.K.; O’Rourke, P.; Wockner, L.F.; deFazio, A.; Butow, P.N.; Price, M.A.; Horwood, K.R.; Clavarino, A.M.; et al. Quality of life and treatment response among women with platinum-resistant versus platinum-sensitive ovarian cancer treated for progression: A prospective analysis. Gynecol. Oncol. 2014, 132, 130–136.
- SEER Ovarian Cancer. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 6 June 2022).
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191.
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res 2016, 9, 27.
- Amirtharaj, F.; Venkatesh, G.H.; Wojtas, B.; Nawafleh, H.H.; Mahmood, A.S.; Nizami, Z.N.; Khan, M.S.; Thiery, J.; Chouaib, S. p53 reactivating small molecule PRIMA-1(MET)/APR-246 regulates genomic instability in MDA-MB-231 cells. Oncol. Rep. 2022, 47, 85.
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67.
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494.
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663.
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775.
- Pelttari, L.M.; Heikkinen, T.; Thompson, D.; Kallioniemi, A.; Schleutker, J.; Holli, K.; Blomqvist, C.; Aittomäki, K.; Bützow, R.; Nevanlinna, H. RAD51C is a susceptibility gene for ovarian cancer. Hum. Mol. Genet. 2011, 20, 3278–3288.
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Eccles, D.; Evans, D.G.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476.
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970.
- Catteau, A.; Harris, W.H.; Xu, C.F.; Solomon, E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: Correlation with disease characteristics. Oncogene 1999, 18, 1957–1965.
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569.
- Bianco, T.; Chenevix-Trench, G.; Walsh, D.C.; Cooper, J.E.; Dobrovic, A. Tumour-specific distribution of BRCA1 promoter region methylation supports a pathogenetic role in breast and ovarian cancer. Carcinogenesis 2000, 21, 147–151.
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487.
- Hurley, R.M.; McGehee, C.D.; Nesic, K.; Correia, C.; Weiskittel, T.M.; Kelly, R.L.; Venkatachalam, A.; Hou, X.; Pathoulas, N.M.; Meng, X.W.; et al. Characterization of a RAD51C-silenced high-grade serous ovarian cancer model during development of PARP inhibitor resistance. NAR Cancer 2021, 3, zcab028.
- Nesic, K.; Kondrashova, O.; Hurley, R.M.; McGehee, C.D.; Vandenberg, C.J.; Ho, G.Y.; Lieschke, E.; Dall, G.; Bound, N.; Shield-Artin, K.; et al. Acquired RAD51C Promoter Methylation Loss Causes PARP Inhibitor Resistance in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2021, 81, 4709–4722.
- Min, A.; Im, S.A.; Yoon, Y.K.; Song, S.H.; Nam, H.J.; Hur, H.S.; Kim, H.P.; Lee, K.H.; Han, S.W.; Oh, D.Y.; et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol. Cancer Ther. 2013, 12, 865–877.
- Goodheart, M.J.; Rose, S.L.; Hattermann-Zogg, M.; Smith, B.J.; De Young, B.R.; Buller, R.E. BRCA2 alteration is important in clear cell carcinoma of the ovary. Clin. Genet. 2009, 76, 161–167.
- Yao, Q.; Liu, Y.; Zhang, L.; Dong, L.; Bao, L.; Bai, Q.; Cui, Q.; Xu, J.; Li, M.; Liu, J.; et al. Mutation Landscape of Homologous Recombination Repair Genes in Epithelial Ovarian Cancer in China and Its Relationship with Clinicopathlological Characteristics. Front. Oncol. 2022, 12, 709645.
- Cao, C.; Yu, R.; Gong, W.; Liu, D.; Zhang, X.; Fang, Y.; Xia, Y.; Zhang, W.; Gao, Q. Genomic mutation features identify distinct BRCA-associated mutation characteristics in endometrioid carcinoma and endometrioid ovarian carcinoma. Aging 2021, 13, 24686–24709.
- Gou, R.; Dong, H.; Lin, B. Application and reflection of genomic scar assays in evaluating the efficacy of platinum salts and PARP inhibitors in cancer therapy. Life Sci. 2020, 261, 118434.
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211.
- Nguyen, L.; WM Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584.
- Sekine, M.; Nishino, K.; Enomoto, T. BRCA Genetic Test and Risk-Reducing Salpingo-Oophorectomy for Hereditary Breast and Ovarian Cancer: State-of-the-Art. Cancers 2021, 13, 2562.
- Ngoi, N.Y.L.; Tan, D.S.P. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144.
- Liu, L.; Matsunaga, Y.; Tsurutani, J.; Akashi-Tanaka, S.; Masuda, H.; Ide, Y.; Hashimoto, R.; Inuzuka, M.; Watanabe, C.; Taruno, K.; et al. BRCAness as a prognostic indicator in patients with early breast cancer. Sci. Rep. 2020, 10, 21173.
- Dhawan, M.; Ryan, C.J. BRCAness and prostate cancer: Diagnostic and therapeutic considerations. Prostate Cancer Prostatic Dis. 2018, 21, 488–498.
- Wong, W.; Raufi, A.G.; Safyan, R.A.; Bates, S.E.; Manji, G.A. BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects. Cancer Manag. Res. 2020, 12, 2731–2742.
- Wang, Y.; Zheng, K.; Huang, Y.; Xiong, H.; Su, J.; Chen, R.; Zou, Y. PARP inhibitors in gastric cancer: Beacon of hope. J. Exp. Clin. Cancer Res. 2021, 40, 211.
- Catalano, F.; Borea, R.; Puglisi, S.; Boutros, A.; Gandini, A.; Cremante, M.; Martelli, V.; Sciallero, S.; Puccini, A. Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer. Cancers 2022, 14, 1388.
- Fritz, C.; Portwood, S.M.; Przespolewski, A.; Wang, E.S. PARP goes the weasel! Emerging role of PARP inhibitors in acute leukemias. Blood Rev. 2021, 45, 100696.
- Jubin, T.; Kadam, A.; Jariwala, M.; Bhatt, S.; Sutariya, S.; Gani, A.R.; Gautam, S.; Begum, R. The PARP family: Insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival. Cell Prolif. 2016, 49, 421–437.
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179.
- Demény, M.A.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions. Cancers 2021, 13, 2057.
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146.
- Abbotts, R.; Dellomo, A.J.; Rassool, F.V. Pharmacologic Induction of BRCAness in BRCA-Proficient Cancers: Expanding PARP Inhibitor Use. Cancers 2022, 14, 2640.
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625.
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25.
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631.
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358.
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246.
- del Rivero, J.; Kohn, E.C. PARP Inhibitors: The Cornerstone of DNA Repair-Targeted Therapies. Oncology 2017, 31, 265–273.
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6.
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Menopause Rev./Przegląd Menopauzalny 2016, 15, 215–219.
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5.
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43.
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years… and Counting. Mol. Cell 2015, 58, 902–910.
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596.
- Benjamin, R.C.; Gill, D.M. ADP-ribosylation in mammalian cell ghosts. Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J. Biol. Chem. 1980, 255, 10493–10501.
- Purnell, M.R.; Whish, W.J. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem. J. 1980, 185, 775–777.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- U.S. Food and Drug Administration. Olaparib. Available online: http://wayback.archive-it.org/7993/20170111231644/http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm427598.htm. (accessed on 8 July 2022).
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261.
- U.S. Food and Drug Administration. Rucaparib. Available online: http://wayback.archive-it.org/7993/20170111231546/http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm533891.htm (accessed on 8 July 2022).
- U.S. Food and Drug Administration. Niraparib (Zejula). Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/niraparib-zejula (accessed on 8 July 2022).
- U.S. Food and Drug Administration. FDA Approves Olaparib Tablets for Maintenance Treatment in Ovarian Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-tablets-maintenance-treatment-ovarian-cancer (accessed on 8 July 2022).
- U.S. Food and Drug Administration. FDA Approves Rucaparib for Maintenance Treatment of Recurrent Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-rucaparib-maintenance-treatment-recurrent-ovarian-fallopian-tube-or-primary-peritoneal (accessed on 8 July 2022).
- FDA. FDA Approves Talazoparib for gBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer. FDA Approves Talazoparib for gBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer|FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-gbrcam-her2-negative-locally-advanced-or-metastatic-breast-cancer (accessed on 22 June 2022).
- Markham, A. Pamiparib: First Approval. Drugs 2021, 81, 1343–1348.
- Lee, A. Fuzuloparib: First Approval. Drugs 2021, 81, 1221–1226.
- U.S. Food and Drug Administration. FDA Approves Niraparib for HRD-Positive Advanced Ovarian Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-hrd-positive-advanced-ovarian-cancer (accessed on 8 July 2022).
- U.S. Food and Drug Administration. FDA approves Olaparib Plus Bevacizumab as Maintenance Treatment for Ovarian, Fallopian Tube, or Primary Peritoneal Cancers. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary (accessed on 8 July 2022).
- Lee, A. Niraparib: A Review in First-Line Maintenance Therapy in Advanced Ovarian Cancer. Target. Oncol. 2021, 16, 839–845.
- U.S. Food and Drug Administration. FDA Approves Niraparib for First-Line Maintenance of Advanced Ovarian Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-first-line-maintenance-advanced-ovarian-cancer (accessed on 22 June 2022).
- Ferraris, D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584.
- Steffen, J.D.; Brody, J.R.; Armen, R.S.; Pascal, J.M. Structural Implications for Selective Targeting of PARPs. Front. Oncol. 2013, 3, 301.
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394.
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.A.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci. Rep. 2020, 10, 2585.
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int. J. Mol. Sci. 2021, 22, 4203.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599.
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477.
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440.
- Navarro, J.; Gozalbo-López, B.; Méndez, A.C.; Dantzer, F.; Schreiber, V.; Martínez, C.; Arana, D.M.; Farrés, J.; Revilla-Nuin, B.; Bueno, M.F.; et al. PARP-1/PARP-2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci. Rep. 2017, 7, 41962.
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419.
- Farrés, J.; Llacuna, L.; Martin-Caballero, J.; Martínez, C.; Lozano, J.J.; Ampurdanés, C.; López-Contreras, A.J.; Florensa, L.; Navarro, J.; Ottina, E.; et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015, 22, 1144–1157.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
12 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No