 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ted Tai-Heng Chen | + 1697 word(s) | 1697 | 2020-11-04 09:34:52 | | | |
| 2 | Lily Guo | Meta information modification | 1697 | 2020-11-05 03:46:59 | | |
Video Upload Options
Facioscapulohumeral muscular dystrophy (FSHD)—the worldwide third most common inherited muscular dystrophy caused by the heterozygous contraction of a 3.3 kb tandem repeat (D4Z4) on a chromosome with a 4q35 haplotype—is a progressive genetic myopathy with variable onset of symptoms, distribution of muscle weakness, and clinical severity. A rough correlation between the phenotypic severity of FSHD and the D4Z4 repeat size has been reported, and the majority of patients with infantile FSHD obtain a very short D4Z4 repeat length (one to three copies, EcoRI size 10–14 kb), in contrast to the classical, slowly progressive, form of FSHD (15–38 kb). With the increasing identifications of case reports and the advance in genetic diagnostics, recent studies have suggested that the variant of FSHD is not a genetically separate entity but a part of the FSHD spectrum. Nevertheless, many questions about the clinical phenotype and natural history of FSHD remain unanswered, limiting evidence-based clinical management.
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD; MIM158900) accounts for one in 15,000–20,000 normal individuals worldwide and is the third most common form of muscular dystrophy after Duchenne and myotonic muscular dystrophy[1]. In 1884, FSHD was first reported by Landouzy and Dejerine[2], who summarized a novel pattern of weakness affecting the facial (facio), shoulder (scapula), and upper arm (humeral) muscles. In 1952, a study of six generations of a family in Utah further described the clinical presentations of FSHD [3]. According to the diagnostic criteria proposed by the International Facioscapulohumeral Consortium[3][4], FSHD is generally defined by the characteristic, slow progression of initially restricted muscle weakness distribution (Table 1). However, because of the high degree of variability in clinical manifestations and the availability of advanced genetic diagnosis, molecular testing has almost replaced these clinical diagnostic criteria for FSHD[5].
Table 1. Diagnostic criteria for facioscapulohumeral dystrophy (FSHD).
|
Inclusion Criteria |
l Facial, humeral, and shoulder girdle muscle weakness (in familial cases, facial weakness is present in >90% of affected individuals) l A weakness of shoulder-girdle muscle groups more prominent than hip-girdle ones (not applicable in severely early-onset cases) l Autosomal dominant inheritance in familial cases |
|
Exclusion Criteria |
l Extraocular or pharyngeal muscle weakness l Prominent and diffuse elbow contractures l Cardiomyopathy l Distal symmetrical sensory loss l Dermatomyositic rash or signs of an alternative diagnosis l Electromyographic evidence of myotonia or neurogenic potentials |
|
Supplementary Criteria |
l Asymmetry of muscle weakness l A descending sequence of involvement l Early, often partial, abdominal muscle weakness (positive Beevor’s sign) l Sparing of deltoid muscles l Typical shoulder profile: straight clavicles, forward sloping of shoulders l Relative sparing of neck flexors l A selective weakness of wrist extensors in distal upper extremities l Sparing of calf muscles l High-frequency hearing loss l Retinal vasculopathy |
Source: Facioscapulohumeral Consortium at the International Conference on the Cause and Treatment of Facioscapulohumeral Dystrophy, Boston, 1997. Data presentation is modified from Liew et al. 2015[3].
2. Genetics and Pathogenesis of FSHD
Two genetically distinct types of FSHD, called FSHD type 1 (FSHD1) and type 2 (FSHD2), are classified. Despite having distinct genotypes, a dysregulated epigenetic repression of the retrogene, called double homeobox 4 (DUX4) in the skeletal muscle, plays a key role in the pathogenesis of both genotypes of FSHD[6]. The DUX4 protein is a transcription factor expressed in the germline, but its complete function is still under investigation. Several pieces of evidence show that DUX4 can regulate the expression of genes associated with stem cell and germline development[7]. Preliminary data suggest that inappropriate expression of DUX4 and its transcriptional targets in skeletal muscle can lead to apoptosis, impaired muscle regeneration (myogenesis), and induce inflammation[5][8]. Besides, DUX4 expression may be associated with clinical severity in FSHD. DUX4 expression, as well as DUX4 target gene dysregulation, can be seen as early as in FSHD-affected fetuses, suggesting that FSHD pathology might occur early in muscle development[9]. Another explanation could be the particular vulnerability to DUX4 of the developing muscles and nervous system during childhood and puberty[10].
FSHD1 is the most common type (95%) of FSHD and accounts for almost all early-onset cases[11]. FSHD1 is a dominantly inherited disease caused by a genetic defect located in the chromosome 4q35 region, where variable deletions occur on a 3.3 kb tandem repeat (D4Z4) array, which can be visualized by EcoRI/Bln1 digested DNA fragment on the Southern blotting gel. In general, healthy individuals have 11 to more than 100 copies (EcoRI 50–300 kb) of D4Z4 repeats, whereas patients with FSHD1 have only ten or fewer D4Z4 repeats (<39 kb) on each copy of 4q35[5][8][12]. Disruption of CpG methylation (hypomethylation), related to contraction of D4Z4 repeats, leads to chromatin relaxation of the 4q35 region, which initiates DUX4 production (Figure 1). Subsequently, the DUX4 expressed from the permissive chromosomal allele with a polyadenylation signal (PAS) results in transcriptional dysregulation and consequent degeneration of muscle cells[2][5]. FSHD2, representing less than 5% of patients with clinically defined FSHD, is not associated with deletions of D4Z4 copies but mutations of the SMCHD1 (on chromosome 18p11) or DNMT3B gene (on chromosome 20q11) in the presence of a disease-permissive 4qA haplotype[6][8][13]. However, the comprehensive molecular genetic testing of FSHD2 is relatively complex and has not been made commercially available so far. Although there are different underlying genetic mechanisms between the two types of FSHD, they cannot be distinguished from clinical features.
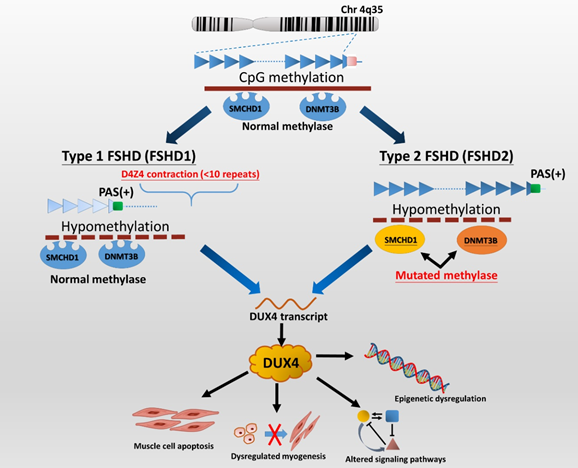
Figure 1. The genetic pathomechanisms of FSHD types 1 and 2. Normal individuals have 11 to more than 100 repeats (50–300 kb) of D4Z4 repeats on the sub-telomeric region of chromosome 4q35, whereas individuals with FSHD1 have a deletion of the array to 10 or fewer D4Z4 units (<39 kb). Contraction of D4Z4 repeats with the presence of a permissive 4qA haplotype or allele containing a polyadenylation signal (PAS, green box) then disrupts CpG methylation (hypomethylation), leading to chromatin relaxation at the array and allowing for the expression of a toxic transcription factor, DUX4 (double homeobox 4). Additionally, the DUX4 retrogene within the D4Z4 unit lacks a stabilizing PAS but can use an additional exon immediately distal to the repeat that contains a polyadenylation signal (DUX4-PAS). In FSHD2, mutation of methylase-related genes, SMCHD1 or DNMT3B, also causes hypomethylation of D4Z4 chromatin structure, resulting in DUX4 expression. There are several potential mechanisms of DUX4 involving in FSHD pathogenies. DUX4 has been implicated as being involved in apoptosis, myogenesis, epigenetic regulation, and regulatory signaling pathways in skeletal muscle and extramuscular tissues, including retinal vessels (Wnt/β-catenin pathway) and auditory cells (JNK pathway)[5][6][11]. Picture modified from Lek et al. 2015[2]and Lim et al. 2020[5].
In the majority of FSHD1 patients, the EcoRI fragment ranges from 15 to 38 kb. In contrast, patients with large contractions of D4Z4 (one to three D4Z4 repeats with EcoRI size of 10–14 kb) usually delineate the most severe phenotype and high frequency of systemic involvements[6][14][15][16]. On the other hand, as many as 30% of FSHD1 cases confirmed by genetic testing may be asymptomatic, especially those with small deletions (8–10 D4Z4 repeats), which delineate the mildest end of the disease severity spectrum[17]. Therefore, it is implied that the fragment size of D4Z4 is roughly negatively correlated with the clinical severity of FSHD[3][8][13][18][19].
3. Clinical Manifestations of FSHD
The onset of FSHD symptoms ranges from birth to 70 years old, and the clinical severity may vary from asymptomatic carriers to patients with extensive muscle wasting, leading to functional dependence and chronic respiratory failure [20]. FSHD is traditionally delineated as muscular dystrophy with slow progression, and the age of onset is variable with a 95% penetrance[3][4]. It usually starts with weakness in the facial, periscapular, and humeral muscle groups, except for the extraocular and deltoid muscles. Muscle weakness usually progresses in a descending manner, gradually involving the trunk, hip girdle, and muscle of distal lower extremities (ankle dorsiflexors). At the advanced stage of FSHD, about 20% (10–30%) of patients become wheelchair-dependent after the age of 50[4][17]. However, around 20–30% of the genetically confirmed cases are asymptomatic, including both FSHD1 and FSHD2 patients[5][21]. These imply a more complex interplay of genetic and epigenetic factors in the manifestation of FSHD phenotypes.
Over time, affected individuals will develop a constellation of signs associated with FSHD, some of which strongly suggest FSHD, whereas other signs might more usually accompany other neuromuscular dystrophies and orthopedic anomalies. Besides, the involvement of other systems has been repeatedly reported in FSHD. Figure 2 illustrates an overview of FSHD-related signs and symptoms, including muscular and extramuscular involvement, in the different body regions. Facial involvement, usually representing the first clinical manifestation, is characterized by eye closure weakness, reduced facial expressions, transverse smile, difficulty in whistling, use of straws, or puckering the lips. The involvement of the shoulder-girdle and humeral muscles can produce the features of scapular winging (high-riding scapula), horizontal clavicles, and limitation in shoulder abduction and elbow flexion. As the disease progresses or onset at a relatively early age, patients with a severely advanced phenotype may exhibit rare features, such as a poly-hill sign, Beevor’s sign, excessive curvature of the lumbar spine (hyperlordosis), Trendelenburg gait as well as drop foot[11][17]. Nevertheless, given that the bulbar, respiratory, and cardiac muscles are relatively unaffected by the disease course, patients with a typical FSHD type usually have a normal life expectancy[3][4][12].
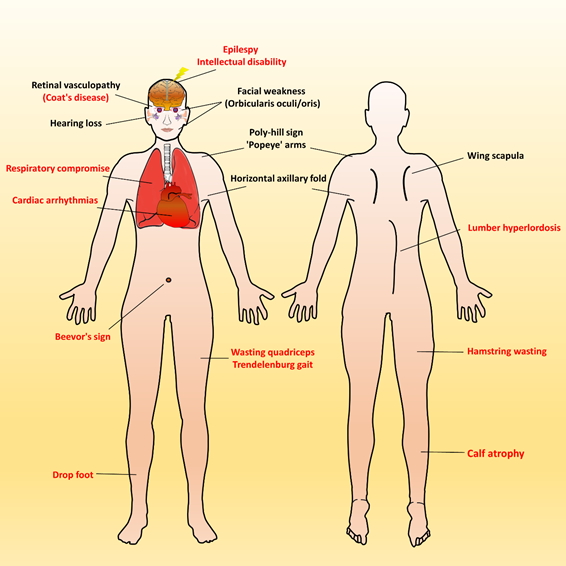
Figure 2. Characteristic muscular and extramuscular signs of FSHD. Generally, most patients with FSHD have multiple distinctive features (as listed in Table 1). Illustrated FSHD-related signs and symptoms marked with red text indicate atypical and relatively rare features that are usually presented in infantile FSHD or at the advanced stage of disease course. Figure modified from Mul et al. 2016[11].
Although FSHD is a hereditary neuromuscular disease, a negative family history certainly does not exclude its diagnosis. A significant percentage of cases, maybe up to 30%, are sporadic de novo mutations, and FSHD families frequently include asymptomatic gene carriers[4][20]. Besides, many patients do not fit this well-known classical FSHD phenotype. Atypical features have been reported, including less facial involvement, neck weakness, calf hypertrophy, and preferential involvement of lower limbs[22][23]. In particular, early-onset and late-onset cases are not uncommon, and the severity and sequence of involvement of different muscle groups may vary. Patients with FSHD may often exhibit non-specific discomforts, such as chronic musculoskeletal pain and fatigue[3][24]. Therefore, if healthcare providers do not actively ask and investigate symptoms and signs, it is easy to miss the diagnosis of FSHD.
References
- Tawil, R. Facioscapulohumeral muscular dystrophy. Curr. Neurol. Neurosci. Rep. 2004, 4, 51–54.
- Lek, A.; Rahimov, F.; Jones, P.L.; Kunkel, L.M. Emerging preclinical animal models for FSHD. Trends Mol. Med. 2015, 21, 295–306.
- Liew, W.K.M.; van der Maarel, S.M.; Tawil, R. Chapter 32—Facioscapulohumeral Dystrophy. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence, 2nd ed.; Darras, B.T., Jones, H.R., Ryan, M.M., De Vivo, D.C., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 620–630.
- Tawil, R.; Kissel, J.T.; Heatwole, C.; Pandya, S.; Gronseth, G.; Benatar, M.; Guideline Development, Dissemination; Implementation Subcommittee of the American Academy of Neurology; Practice Issues Review Panel of the American Association of Neuromuscular; Electrodiagnostic Medicine. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 85, 357–364.
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729.
- Statland, J.M. Chapter 30—Facioscapulohumeral muscular dystrophy. In Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: San Diego, CA, USA, 2020; pp. 511–523.
- Daxinger, L.; Tapscott, S.J.; van der Maarel, S.M. Genetic and epigenetic contributors to fshd. Curr. Opin. Genet. Dev. 2015, 33, 56–61.
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genom. Hum. Genet. 2019, 20, 265–291.
- Ferreboeuf, M.; Mariot, V.; Bessieres, B.; Vasiljevic, A.; Attie-Bitach, T.; Collardeau, S.; Morere, J.; Roche, S.; Magdinier, F.; Robin-Ducellier, J.; et al. Dux4 and dux4 downstream target genes are expressed in fetal fshd muscles. Hum. Mol. Genet. 2014, 23, 171–181.
- Goselink, R.J.M.; Mul, K.; van Kernebeek, C.R.; Lemmers, R.; van der Maarel, S.M.; Schreuder, T.H.A.; Erasmus, C.E.; Padberg, G.W.; Statland, J.M.; Voermans, N.C.; et al. Early onset as a marker for disease severity in facioscapulohumeral muscular dystrophy. Neurology 2019, 92, e378–e385.
- Mul, K.; Lassche, S.; Voermans, N.C.; Padberg, G.W.; Horlings, C.G.; van Engelen, B.G. What’s in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pr. Neurol. 2016, 16, 201–207.
- Hamel, J.; Tawil, R. Facioscapulohumeral muscular dystrophy: Update on pathogenesis and future treatments. Neurotherapeutics 2018, 15, 863–871.
- Magdinier, F.; Upadhyaya, M. Facioscapulohumeral Muscular Dystrophy: Genetics; eLS John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 1–13.
- Trevisan, C.P.; Pastorello, E.; Tomelleri, G.; Vercelli, L.; Bruno, C.; Scapolan, S.; Siciliano, G.; Comacchio, F. Facioscapulohumeral muscular dystrophy: Hearing loss and other atypical features of patients with large 4q35 deletions. Eur. J. Neurol. 2008, 15, 1353–1358.
- Nikolic, A.; Ricci, G.; Sera, F.; Bucci, E.; Govi, M.; Mele, F.; Rossi, M.; Ruggiero, L.; Vercelli, L.; Ravaglia, S.; et al. Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1–3 d4z4 reduced alleles: Experience of the fshd italian national registry. BMJ Open 2016, 6, e007798.
- Chen, T.H.; Lai, Y.H.; Lee, P.L.; Hsu, J.H.; Goto, K.; Hayashi, Y.K.; Nishino, I.; Lin, C.W.; Shih, H.H.; Huang, C.C.; et al. Infantile facioscapulohumeral muscular dystrophy revisited: Expansion of clinical phenotypes in patients with a very short ecori fragment. Neuromuscul. Disord. 2013, 23, 298–305.
- Mah, J.K.; Chen, Y.W. A pediatric review of facioscapulohumeral muscular dystrophy. J. Pediatr. Neurol. 2018, 16, 222–231.
- Ruggiero, L.; Mele, F.; Manganelli, F.; Bruzzese, D.; Ricci, G.; Vercelli, L.; Govi, M.; Vallarola, A.; Tripodi, S.; Villa, L.; et al. Phenotypic variability among patients with d4z4 reduced allele facioscapulohumeral muscular dystrophy. JAMA Netw. Open 2020, 3, e204040.
- Steel, D.; Main, M.; Manzur, A.; Muntoni, F.; Munot, P. Clinical features of facioscapulohumeral muscular dystrophy 1 in childhood. Dev. Med. Child Neurol. 2019, 61, 964–971.
- Goselink, R.J.M.; Voermans, N.C.; Okkersen, K.; Brouwer, O.F.; Padberg, G.W.; Nikolic, A.; Tupler, R.; Dorobek, M.; Mah, J.K.; van Engelen, B.G.M.; et al. Early onset facioscapulohumeral dystrophy—A systematic review using individual patient data. Neuromuscul. Disord. 2017, 27, 1077–1083.
- Mah, J.K.; Feng, J.; Jacobs, M.B.; Duong, T.; Carroll, K.; de Valle, K.; Carty, C.L.; Morgenroth, L.P.; Guglieri, M.; Ryan, M.M.; et al. A multinational study on motor function in early-onset fshd. Neurology 2018, 90, e1333–e1338.
- Rijken, N.H.; van der Kooi, E.L.; Hendriks, J.C.; van Asseldonk, R.J.; Padberg, G.W.; Geurts, A.C.; van Engelen, B.G. Skeletal muscle imaging in facioscapulohumeral muscular dystrophy, pattern and asymmetry of individual muscle involvement. Neuromuscul. Disord. 2014, 24, 1087–1096.
- Pastorello, E.; Cao, M.; Trevisan, C.P. Atypical onset in a series of 122 cases with facioscapulohumeral muscular dystrophy. Clin. Neurol. Neurosurg. 2012, 114, 230–234.
- Voet, N.; Bleijenberg, G.; Hendriks, J.; de Groot, I.; Padberg, G.; van Engelen, B.; Geurts, A. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in fshd: An rct. Neurology 2014, 83, 1914–1922.

