 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qing Zhang | + 1809 word(s) | 1809 | 2020-11-02 10:28:59 |
Video Upload Options
The oxygen-sensing system is a complicated and elaborate system containing large molecular components, including hypoxia-inducible factors (HIFs) as the central regulator of oxygen homeostasis, prolyl hydroxylases (PHDs), Von Hippel-Lindau protein (pVHL) as the modulator of ubiquitin-mediated proteolysis, and the co-factors and downstream targets as functional contributors. It should be recognized that the PHDs-HIF-pVHL axis remains the best-characterized and central signaling in the oxygen sensing pathway, although novel mechanisms continue to be illuminated. In this entry, we summarize the current knowledge about canonical hypoxia signaling, including the function of HIF transcription factors, prolyl hydroxylation of HIFs, and pVHL.
1. HIF Transcription Factors: The Central Regulator of Oxygen Homeostasis
The capacity to recognize and adapt to variations in oxygen is meaningful for cellular and whole-organism homeostasis and survival. Nearly all mammalian cells respond to reduced oxygen availability by controlling the transcription factor HIF [1]. Of the 3 encoded HIF isoforms in humans (i.e., HIF-1, HIF-2, and HIF-3), the transcriptional responses to HIF-1 and HIF-2 isoforms, specifically, have been the most studied.
HIF-1 is a heterodimer of basic-helix-loop-helix-Per-ARNT-Sim proteins-HIF-1α (encoded by HIF1A) and HIF-1β (encoded by aryl hydrocarbon receptor nuclear translocator or ARNT)[2]. HIF-1α mediates the HIF complex’s oxygen sensitivity and interacts with HIF-1β by an authorized Per–ARNT–Sim (PAS) domain [3]. Both HIF-1α and HIF-1β comprise an N-terminal basic-helix-loop-helix domain responsible for DNA binding and C-terminal transactivation domains which induce gene expression[3]. The HIF-2α and HIF-3α complexes with HIF-1β are known as HIF-2 and HIF-3 respectively. HIF-α proteins are oxygen-sensitive in that they contain an oxygen-dependent degradation (ODD) domain with target prolyl residues whose hydroxylation mediates interaction with pVHL, and also the C-terminal transactivation domain, which contains the target asparaginyl residue [3][4][5] (Figure 1). When oxygen is present, hydroxylation of the prolyl or asparaginyl residue by a prolyl hydroxylase or factor-inhibiting HIF (FIH) results in pVHL binding and subsequent HIF proteasome-dependent degradation. Under hypoxic conditions, pVHL-mediated degradation does not occur, and HIF-1α stabilization allows the generation of the HIF-1 heterodimer, which is translocated to the nucleus and occupies hypoxia-responsive elements (HREs)-containing gene promoters and their transcriptional enhancers [3]. Human HIF-1 has been identified as a protein complex binding to a regulatory DNA sequence at the EPO locus [6]. Cooperation with other DNA-binding proteins can coactivate HIF, fine-tuning HIF targets [7][8].
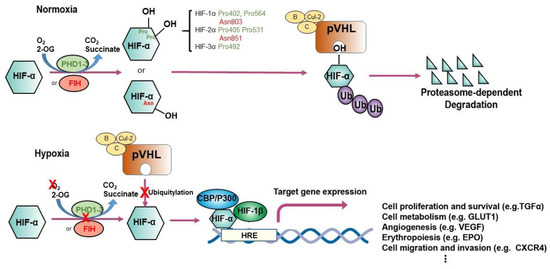
Figure 1. Regulation of hypoxia-inducible factor (HIF)-1α and HIF-2α. The schematic illustrates different modes of regulation of HIF-1α and HIF-2α. Under conditions of normoxia, prolyl hydroxylation regulated by EglN1-3 or asparaginyl hydroxylation regulated by factor inhibiting HIF (FIH) promote binding of HIF-α subunits to the von Hippel–Lindau (VHL) tumor suppressor protein (pVHL), the recognition component of a ubiquitin E3 ligase complex; ubiquitylation (Ub) targets HIF for proteasomal degradation. Under hypoxic conditions, the hydroxylation of HIF-α is inhibited, permitting interaction with the acetyltransferases p300 and CREB-binding protein (CBP) and further increasing transcription of HIF target genes. EPO, erythropoietin; HRE, hypoxia-responsive element, VEGF, vascular endothelial growth factor; TGF-α, transforming growth factor α; GLUT1, glucose transporter 1.
HIF-1 and HIF-2 both transduce positive transcriptional responses to hypoxia, although their transcriptional targets, the kinetics of activation, and oxygen dependence differ [3][9][10]. These variations might partly be attributed to different transcriptional feedback circles. For example, an antisense HIF transcript might negatively modulate HIF-1α expression by destabilizing HIF-1α mRNA [11]. Despite binding to an identical DNA sequence at HREs, HIF-1 and HIF-2 may direct largely distinct transcriptional systems [12][13][14][15]. For instance, many metabolic responses are dependent on HIF-1, whereas cell differentiation, reparative pathways, and more complex adaptive responses to hypoxia, including induction of erythropoiesis, are dependent on HIF-2 [16][17][18]. Interestingly, HIF-1 and HIF-2 also have different patterns of genomic distribution concerning their transcriptional targets. While HIF-1 generally binds to DNA at sites in close proximity to target gene promoters, HIF-2 frequently binds to transcriptional enhancers that lie at a distance from the target gene promoter [12], and this pattern is maintained across cell types with quite different complements of HIF target genes[19]. Although some HREs can be recognized by both HIF-1 and HIF-2, very little cross-competition is observed, so that a HRE can be bound by only one HIF. The two isoforms have distinct chromatin binding patterns [19], which may be connected post-DNA binding mechanisms that mediate transcriptional selectivity [20][21][22]. Together with cell-type-specific expression of HIF-α isoforms, DNA binding selectivity generates highly distinct functional outputs for HIF-1 and HIF-2.
To date, investigation of the human transcriptional targets and tissue/cell-type specificity of HIF-3 expression has been much less complete since there are several splicing isoforms of HIF-3A gene products [23]. Some studies report that HIF-3α might negatively regulate HIF-1α and HIF-2α [24]. For instance, a splice variant, inhibitory PAS protein domain (IPAS), competitively binds to HIF-1/2α, preventing binding to DNA and inhibiting the HIF-1-mediated transcriptional response [25][26]. On the other hand, a study on zebrafish Hif-3α, an orthologue to HIF-3, reported that overexpressed Hif-3α directs an extensive positive transcriptional response [27]. Furthermore, of more than 150 genes that were found to be upregulated by Hif-3α in zebrafish embryos, almost 100 were also Hif-1 targets. These experiments also suggest that Hif-3α might preferentially target specific hypoxia pathways, such as the Janus kinase (JAK)–STAT and NOD-like receptor (NLR) signaling, and that at least some of the targets of zebrafish Hif-3 might also be responsive to HIF-3 in human cells [27].
Genes controlled directly or indirectly by HIF mediate an extensive spectrum of biological outputs. At the cellular level, responses to HIF include effects on differentiation, migration, cytoprotection, apoptosis, cycle control, and mitochondrial function [28][29][30]. At the organ or organism level, coordinated responses to altered HIF activity include metabolic regulation, erythropoiesis, angiogenesis, tissue remodeling, and wound healing [31][32][33][34]. The aforementioned transcriptional adaptations enable normal cells or tissues to survive in a severely oxygen-deficient environment. On the other hand, in the context of cancer, these processes can facilitate tumor progression [35]. Distinctly, in solid tumors, certain regions usually undergo hypoxic status due to abnormal blood vessel development[36]. Accordingly, this was accompanied by elevated HIF-α levels in many solid tumors. HIF activation remodels cellular metabolic oxidative mechanisms, enabling cells to mitigate toxic reactive oxygen species and preserve the synthesis of macromolecules in response to oxygen availability. Hypoxia and accumulation of HIF is also involved in resistance to chemotherapy and radiotherapy and worse prognosis in cancer patients [35]. The HIF-mediated reprogramming of varied and numerous cellular processes such as metabolism, proliferation, angiogenesis, metastasis, invasion, epithelial–mesenchymal transition (EMT), anti-apoptosis, growth factor signaling, and stem cell maintenance [35][37][38][39][40][41][42][43], underscores the significant role of HIF in cancer progression and tumorigeneses.
2. Prolyl Hydroxylation: The Adaptive Mediator of HIFs
Identification of HIF-α proteins as the oxygen-dependent subunits of HIF prompted subsequent efforts to define the molecular mechanisms of oxygen sensing upstream of the HIF signal transduction pathway [3]. As a first step, domains within the HIF-α polypeptide that could confer oxygen-regulated instability onto heterologous proteins were characterized [44][45]. Surprisingly, these domains did not seem to be regulated by protein phosphorylation pathways, which had been widely predicted to transduce the oxygen-sensitive signal [46].
In the presence of oxygen, HIF-α is trans-4-hydroxylated at prolyl residues (Pro402 and Pro564 in HIF-1α; Pro405 and Pro531 in HIF-2α; Pro492 in HIF-3α) [47][48], resulting in a >1000-fold increase in affinity for pVHL. Further analyses identified pVHL as the recognition component of an E3 ubiquitin ligase complex that targets HIF-α for proteasomal destruction by binding to the domains of HIF-α that confer oxygen-regulated instability [49][50]. pVHL is required for the oxygen-dependent proteolysis of HIF-α [51]. Post-translational hydroxylation of specific amino acid residues in HIF-α is the critical oxygen-regulated event promoting HIF-pVHL association [47][48]. Mechanistically, prolyl hydroxylation of HIF-α is coupled to the oxidative decarboxylation of prolyl hydroxylase creating a ferryl intermediate at the catalytic site [3]. HIF prolyl hydroxylation and degradation are highly efficient, so that HIF is very labile and barely detectable in normoxia. As oxygen level decreases, prolyl hydroxylation diminishes, thus enabling HIF levels to rise and the hypoxic response to be “switched on.”
3. pVHL: The Proteolysis Modulator of HIFs
The physiological and pathological function of pVHL has constantly been explored since the first description of VHL disease in the early 1990s by William G. Kaelin Jr. and colleagues [52]. VHL syndrome is an autosomal-dominant, hereditary neoplastic disease associated with clear cell renal cell carcinoma (ccRCC), central nervous system hemangioblastomas, and pheochromocytoma [53]. The disease is caused by germline mutations in VHL, a tumor-suppressor gene located on chromosome 3p25.1 [54]. Patients who inherit a single faulty copy of VHL develop the disease only after spontaneous inactivation or loss of the second, wild-type VHL allele. The leading cause of death in patients with VHL disease is ccRCC [53]. The product of VHL, pVHL, is a 30 kDa protein with multiple functions. The best-documented of these functions relates to its role as the substrate-recognition component of an E3 ubiquitin ligase complex that also contains elongin B, elongin C, cullin-2, and RING-box protein 1 [49][52][55][56][57][58]. This complex is best known for its ability to target HIFαs for polyubiquitination and proteasomal degradation[49][59][60] (Figure 1), although it has been reported to target other substrates including atypical protein kinase C, N-Myc downstream-regulated gene 3 (NDRG3), Akt, erythropoietin receptor (EPOR), transcription factor B-Myb, actin cross-linker filamin A (FLNA), centrosomal protein 68 (CEP68), ceramide kinase-like protein (CERKL), and hsRPB7 [61][62][63][64][65]. In addition, by developing an in vitro VHL-capture-based binding assay combined with a genome-wide screening strategy, we have demonstrated that the zinc fingers and homeoboxes 2 (ZHX2) and the Scm-like with four malignant brain tumor domains 1 (SFMBT1) transcription factors served as novel pVHL substrates in ccRCC [66][67].
An increasing amount of evidence indicates the existence of HIF-independent pathways in the VHL-supervised background, as the simple inhibition of the HIF system might not be sufficient to prevent tumor progression. Therefore, the discovery of additional pVHL targets is critical for the development of new therapeutics. In ccRCC, a novel pVHL substrate, TANK binding kinase 1 (TBK1), an essential kinase involved in the innate immune response, was recently discovered [68]. We found that VHL-deficient kidney tumors display elevated TBK1 phosphorylation. Through genetic ablation, pharmacologic inhibition, or new carbon-based proteolysis targeting chimera specifically, TBK1 was depleted/inhibited in vitro, suppressing VHL-deficient kidney cancer cell proliferation while having no effect on VHL wild-type cells. Similarly, TBK1 depletion abrogates kidney tumorigenesis in an orthotopic xenograft tumor model [68]. Mechanistically, hydroxylation on Pro48 of TBK1 triggers both pVHL and phosphatase PPM1B binding, leading to TBK1 de-phosphorylation. TBK1 contributes to kidney cancer cell growth by phosphorylating p62/SQSTM1 on Ser366, thus stabilizing p62. In this way, TBK1, distinct from its innate immune signaling role, might serve as a novel target with therapeutic potential for cancers with VHL loss.
It is essential to note that pVHL targets other proteins in an E3 ubiquitin-ligase-independent manner with or without oxygen signal involvement. Proteins subjected to this regulatory fashion by pVHL are, for example, TBK1, aldehyde dehydrogenase 2 (ALDH2), p53, AKT, and Caspase Recruitment Domain Family Member 9 (Card9) [68][69][70][71][72]. In summary, these findings highlight the critical role of pVHL in the oxygen-signaling pathway and the control of the abundance or activity of its substrates in the context of disease.
References
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361, doi:10.1002/wsbm.69
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514, doi:10.1073/pnas.92.12.5510.
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659, doi:10.1038/s41581-019-0182-z.
- Kaelin, W.G., Jr. The von Hippel–Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873, doi:10.1038/nrc2502.
- Okumura, F.; Uematsu, K.; Byrne, S.D.; Hirano, M.; Joo-Okumura, A.; Nishikimi, A.; Shuin, T.; Fukui, Y.; Nakatsukasa, K.; Kamura, T. Parallel Regulation of von Hippel-Lindau Disease by pVHL-Mediated Degradation of B-Myb and Hypoxia-Inducible Factor α. Mol. Cell. Biol. 2016, 36, 1803–1817, doi:10.1128/mcb.00067-16.
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454, doi:10.1128/mcb.12.12.5447.
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide Association of Hypoxia-inducible Factor (HIF)-1α and HIF-2α DNA Binding with Expression Profiling of Hypoxia-inducible Transcripts. J. Biol. Chem. 2009, 284, 16767–16775, doi:10.1074/jbc.m901790200.
- Jiang, B.-H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA Binding, and Transactivation Properties of Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 17771–17778, doi:10.1074/jbc.271.30.17771.
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Åke; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423, doi:10.1016/j.ccr.2006.08.026.
- Rosenberger, C.; Mandriota, S.; Jürgensen, J.S.; Wiesener, M.S.; Hörstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.-U. Expression of Hypoxia-Inducible Factor-1 and -2 in Hypoxic and Ischemic Rat Kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732, doi:10.1097/01.asn.0000017223.49823.2a.
- Rossignol, F.; Vaché, C.; Clottes, E. Natural antisense transcripts of hypoxia-inducible factor 1alpha are detected in different normal and tumour human tissues. Gene 2002, 299, 135–140, doi:10.1016/s0378-1119(02)01049-1.
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217, doi:10.1182/blood-2010-10-314427.
- Greenald, D.; Jeyakani, J.; Pelster, B.; Sealy, I.; Mathavan, S.; Van Eeden, F.J. Genome-wide mapping of Hif-1α binding sites in zebrafish. BMC Genom. 2015, 16, 923, doi:10.1186/s12864-015-2169-x.
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Camenisch, G.; Gassmann, M. Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. JBIC J. Biol. Inorg. Chem. 1998, 253, 771–777, doi:10.1046/j.1432-1327.1998.2530771.x.
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567, doi:10.1101/gr.147942.112.
- Hu, C.-J.; Iyer, S.; Sataur, A.; Covello, K.L.; Chodosh, L.A.; Simon, M.C. Differential Regulation of the Transcriptional Activities of Hypoxia-Inducible Factor 1 Alpha (HIF-1α) and HIF-2α in Stem Cells. Mol. Cell. Biol. 2006, 26, 3514–3526, doi:10.1128/mcb.26.9.3514-3526.2006.
- Warnecke, C.; Zaborowska, Z.; Kurreck, J.; Erdmann, V.A.; Frei, U.; Wiesener, M.; Eckardt, K. Differentiating the functional role of hypoxia‐inducible factor (HIF)‐1α and HIF‐2α (EPAS‐1) by the use of RNA interference: Erythropoietin is a HIF‐2α target gene in Hep3B and Kelly cells. FASEB J. 2004, 18, 1462–1464, doi:10.1096/fj.04-1640fje.
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2 regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570, doi:10.1101/gad.1399906.
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA ‐binding specificities of the HIF ‐1α and HIF ‐2α transcription factors in chromatin. EMBO Rep. 2018, 20, doi:10.15252/embr.201846401.
- Platt, J.L.; Salama, R.; Smythies, J.; Choudhry, H.; Davies, J.; Hughes, J.R.; Ratcliffe, P.J.; Mole, D.R. Capture‐C reveals preformed chromatin interactions between HIF ‐binding sites and distant promoters. EMBO Rep. 2016, 17, 1410–1421, doi:10.15252/embr.201642198.
- Lau, K.W.; Tian, Y.-M.; Raval, R.R.; Ratcliffe, P.J.; Pugh, C.W. Target gene selectivity of hypoxia-inducible factor-α in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br. J. Cancer 2007, 96, 1284–1292, doi:10.1038/sj.bjc.6603675.
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-Terminal Transactivation Domain Confers Target Gene Specificity of Hypoxia-inducible Factors HIF-1α and HIF-2α. Mol. Biol. Cell 2007, 18, 4528–4542, doi:10.1091/mbc.e06-05-0419.
- Ravenna, L.; Salvatori, L.; Russo, M.A. HIF3α: The little we know. FEBS J. 2015, 283, 993–1003, doi:10.1111/febs.13572.
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and Characterization of Hypoxia-Inducible Factor (HIF)-3α in Human Kidney: Suppression of HIF-Mediated Gene Expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813, doi:10.1006/bbrc.2001.5659.
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS Domain Protein (IPAS) Is a Hypoxia-inducible Splicing Variant of the Hypoxia-inducible Factor-3α Locus. J. Biol. Chem. 2002, 277, 32405–32408, doi:10.1074/jbc.c200328200.
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186, doi:10.1016/j.yexcr.2017.03.013.
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.-J.; Duan, C. Hypoxia-Inducible Factor 3 Is an Oxygen-Dependent Transcription Activator and Regulates a Distinct Transcriptional Response to Hypoxia. Cell Rep. 2014, 6, 1110–1121, doi:10.1016/j.celrep.2014.02.011.
- Lee, K.E.; Simon, M.C. From stem cells to cancer stem cells: HIF takes the stage. Curr. Opin. Cell Biol. 2012, 24, 232–235, doi:10.1016/j.ceb.2012.01.005.
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. et Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1263–1268, doi:10.1016/j.bbamcr.2010.08.006.
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Physiol. 2015, 309, C775–C782, doi:10.1152/ajpcell.00279.2015.
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408, doi:10.1016/j.cell.2012.01.021.
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79, doi:10.1161/CIRCRESAHA.117.305109.
- Masson, N.; Ratcliffe, P.J. Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014, 2, 3, doi:10.1186/2049-3002-2-3.
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immun. 2014, 41, 518–528, doi:10.1016/j.immuni.2014.09.008.
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214, doi:10.1016/j.tips.2012.01.005.
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451, doi:10.1172/jci120855.
- 63. Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer research 1999, 59, 5830–5835.
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603, doi:10.1371/journal.pone.0129603.
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056.
- Carroll, V.A.; Ashcroft, M. Role of Hypoxia-Inducible Factor (HIF)-1α versus HIF-2α in the Regulation of HIF Target Genes in Response to Hypoxia, Insulin-Like Growth Factor-I, or Loss of von Hippel-Lindau Function: Implications for Targeting the HIF Pathway. Cancer Res. 2006, 66, 6264–6270, doi:10.1158/0008-5472.can-05-2519.
- Yu, E.Z.; Li, Y.-Y.; Liu, X.-H.; Kagan, E.; McCarron, R.M. Antiapoptotic action of hypoxia-inducible factor-1α in human endothelial cells. Lab. Investig. 2004, 84, 553–561, doi:10.1038/labinvest.3700071.
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56, doi:10.1016/j.gde.2009.10.009.
- Volm, M.; Koomägi, R. Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer. Res. 2000, 20, 1527–1533.
- Srinivas, V.; Zhang, L.-P.; Zhu, X.-H.; Caro, J. Characterization of an Oxygen/Redox-Dependent Degradation Domain of Hypoxia-Inducible Factor α (HIF-α) Proteins. Biochem. Biophys. Res. Commun. 1999, 260, 557–561, doi:10.1006/bbrc.1999.0878.
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and Inhibitory Domains of Hypoxia-inducible Factor 1α: Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260, doi:10.1074/jbc.272.31.19253.
- Pugh, C.W.; O’Rourke, J.F.; Nagao, M.; Gleadle, J.M.; Ratcliffe, P.J. Activation of Hypoxia-inducible Factor-1; Definition of Regulatory Domains within the α Subunit. J. Biol. Chem. 1997, 272, 11205–11214, doi:10.1074/jbc.272.17.11205.
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206, doi:10.1093/emboj/20.18.5197.
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1 binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635, doi:10.1073/pnas.181341498.
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia Inducible Factor-α Binding and Ubiquitylation by the von Hippel-Lindau Tumor Suppressor Protein. J. Biol. Chem. 2000, 275, 25733–25741, doi:10.1074/jbc.m002740200.
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472, doi:10.1126/science.1059796.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275, doi:10.1038/20459.
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446, doi:10.1126/science.7660130.
- Maher, E.R.; Yates, J.R.W.; Harries, R.; Benjamin, C.; Moore, A.T.; Ferguson-Smith, M.A.; Harris, R. Clinical Features and Natural History of von Hippel-Lindau Disease. Qjm: Int. J. Med. 1990, 77, 1151–1163, doi:10.1093/qjmed/77.2.1151.
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320, doi:10.1126/science.8493574.
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.-Y.; Huang, L.E.; Pavletich, N.P.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427, doi:10.1038/35017054.
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-la by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309.
- Kaelin, W.G. The von Hippel–Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 2005, 338, 627–638, doi:10.1016/j.bbrc.2005.08.165.
- Kaelin, W.G. The von Hippel‐Lindau Tumor Suppressor Protein: An Update; Academic Press: Waltham, MA, 2007; Vol. 435, pp. 371–383.
- Nyhan, M.J.; O’Sullivan, G.C.; McKenna, S.L. Role of the VHL (von Hippel–Lindau) gene in renal cancer: A multifunctional tumour suppressor. Biochem. Soc. Trans. 2008, 36, 472–478, doi:10.1042/bst0360472.
- Kaelin, W.G., Jr.; Von Hippel-Lindau Disease. Annual Review of Pathology. 2007, 2, 145–173.
- Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, 35, doi:10.3390/biomedicines6010035.
- Yin, H.; Zheng, L.; Liu, W.; Zhang, D.; Li, W.; Yuan, L. Rootletin prevents Cep68 from VHL-mediated proteasomal degradation to maintain centrosome cohesion. Biochim. et Biophys. Acta (BBA) Bioenerg. 2017, 1864, 645–654, doi:10.1016/j.bbamcr.2017.01.007.
- Okuda, H.; Saitoh, K.; Hirai, S.-I.; Iwai, K.; Takaki, Y.; Baba, M.; Minato, N.; Ohno, S.; Shuin, T. The von Hippel-Lindau Tumor Suppressor Protein Mediates Ubiquitination of Activated Atypical Protein Kinase C. J. Biol. Chem. 2001, 276, 43611–43617, doi:10.1074/jbc.m107880200.
- Lai, Y.; Qiao, M.; Song, M.; Weintraub, S.T.; Shiio, Y. Quantitative Proteomics Identifies the Myb-Binding Protein p160 as a Novel Target of the von Hippel-Lindau Tumor Suppressor. PLoS ONE 2011, 6, e16975, doi:10.1371/journal.pone.0016975.
- Na, X.; Duan, H.O.; Messing, E.M.; Schoen, S.R.; Ryan, C.K.; Di Sant’Agnese, P.; Golemis, E.A.; Wu, G. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel–Lindau protein. EMBO J. 2003, 22, 4249–4259, doi:10.1093/emboj/cdg410.
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.-D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295, doi:10.1126/science.aap8411.
- Liu, X.; Simon, J.M.; Xie, H.; Hu, L.; Wang, J.; Zurlo, G.; Fan, C.; Ptacek, T.S.; Herring, L.; Tan, X.; et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol. Cell 2020, 77, 1294–1306.e1295, doi:10.1016/j.molcel.2020.01.009.
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C.; et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2019, 10, 460–475, doi:10.1158/2159-8290.cd-19-0837.
- Gao, Y.-H.; Wu, Z.-X.; Xie, L.-Q.; Li, C.-X.; Mao, Y.-Q.; Duan, Y.-T.; Han, B.; Han, S.-F.; Yu, Y.; Lu, H.-J.; et al. VHL deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating ALDH2. Nat. Commun. 2017, 8, 15337, doi:10.1038/ncomms15337.
- Roe, J.-S.; Youn, H.-D. The Positive Regulation of p53 by the Tumor Suppressor VHL. Cell Cycle 2006, 5, 2054–2056, doi:10.4161/cc.5.18.3247.
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932, doi:10.1126/science.aad5755.
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G. pVHL Acts as an Adaptor to Promote the Inhibitory Phosphorylation of the NF-κB Agonist Card9 by CK2. Mol. Cell 2007, 28, 15–27, doi:10.1016/j.molcel.2007.09.010.

