Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ava Boutilier | -- | 2192 | 2022-10-08 02:29:55 | | | |
| 2 | Dean Liu | Meta information modification | 2192 | 2022-10-08 03:22:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Boutilier, A.J.; Huang, L.; Elsawa, S.F. Mechanisms of Disease Progression of Waldenström Macroglobulinemia. Encyclopedia. Available online: https://encyclopedia.pub/entry/28371 (accessed on 25 July 2026).
Boutilier AJ, Huang L, Elsawa SF. Mechanisms of Disease Progression of Waldenström Macroglobulinemia. Encyclopedia. Available at: https://encyclopedia.pub/entry/28371. Accessed July 25, 2026.
Boutilier, Ava J., Lina Huang, Sherine F. Elsawa. "Mechanisms of Disease Progression of Waldenström Macroglobulinemia" Encyclopedia, https://encyclopedia.pub/entry/28371 (accessed July 25, 2026).
Boutilier, A.J., Huang, L., & Elsawa, S.F. (2022, October 08). Mechanisms of Disease Progression of Waldenström Macroglobulinemia. In Encyclopedia. https://encyclopedia.pub/entry/28371
Boutilier, Ava J., et al. "Mechanisms of Disease Progression of Waldenström Macroglobulinemia." Encyclopedia. Web. 08 October, 2022.
Copy Citation
Waldenström macroglobulinemia is an indolent, B-cell lymphoma without a known cure. The bone marrow microenvironment and cytokines both play key roles in Waldenström macroglobulinemia (WM) tumor progression. Only one FDA-approved drug exists for the treatment of WM, Ibrutinib, but treatment plans involve a variety of drugs and inhibitors.
Waldenström macroglobulinemia
B-cell lymphoma
non-Hodgkin’s
1. Proliferation
IL-21 is a type I cytokine commonly found in the WM tumor microenvironment that rapidly induces the phosphorylation of STAT3 in WM cells (Figure 1) [1]. MWCL-1 cells cultured in the presence of IL-21 for 72 h in vitro demonstrated a dose-dependent increase in both WM cell proliferation and phosphorylated STAT3 levels in those cells [1]. Additionally, in MWCL-1 cells, 10 min of stimulation with IL-21 displayed a significant increase in the phosphorylation of STAT3 [1]. Treatment with a STAT3 inhibitor eliminated the IL-21-mediated increase in proliferation [1].
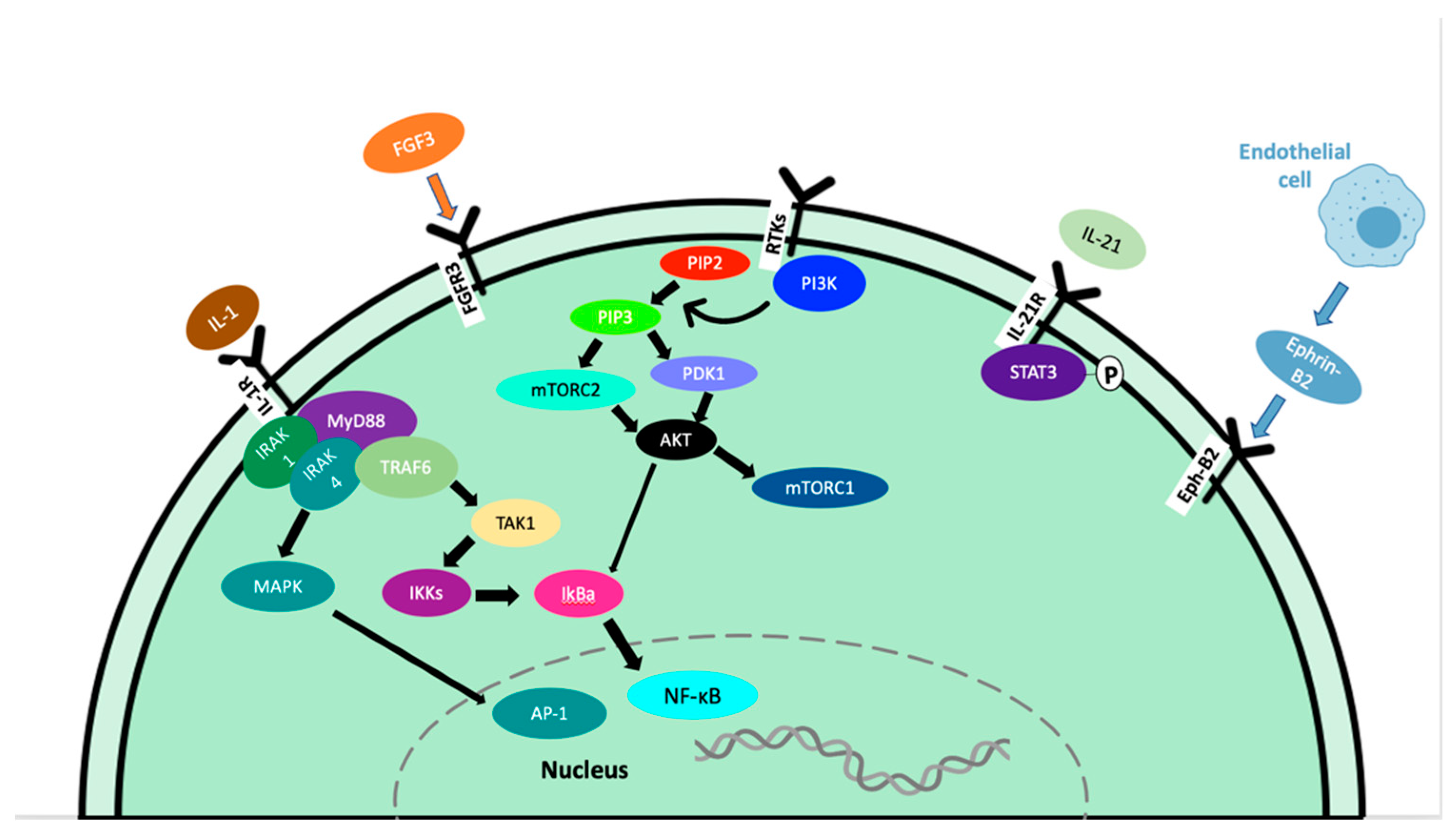
Figure 1. Signaling pathways contributing to tumor progression in WM.
Fibroblast growth factor receptor 3 (FGFR3) is a member of the FGFR family that interacts with fibroblast growth factor 3 (FGF3), inducing a cascade of downstream signals that influence cell proliferation. This is well documented in many types of cancer, including tongue, colorectal, breast, bladder, and oral cancers [2][3][4][5][6][7]. In WM, the expression of FGFR3 on CD19+ cells from WM patients was greater than the expression on B cells from healthy subjects, and FGFR3 was also overexpressed in the cell lines BCWM.1 and MEC-1 [8].
In cancer, overexpression of the Akt and mTOR pathways play an important role in the progression of malignancies through the phosphatidylinositol-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway. This pathway can enhance cell survival by inhibiting cell death and stimulating cell proliferation [9][10]. The activation of this pathway ultimately leads to growth, angiogenesis, resistance to apoptosis, and therapy resistance [11][12]. In WM, constitutive activation of the PI3K/Akt pathway exists and leads to increased cell proliferation and resistance to apoptosis [13]. Phosphatases and tensin homolog (PTEN) are haploinsufficient tumor suppressors; therefore, partial loss-of-function mutations can have a dramatic effect on cancer progression. PTEN acts to deactivate the PI3K/Akt/mTOR pathways, therefore loss-of-function can lead to constitutive activation. Studies in mouse models have shown that even a small reduction in PTEN expression can significantly increase cancer risk [14][15]. Unfortunately, PTEN loss-of-function mutations are frequent in human cancers, leading to the perpetual activation of AKT. Furthermore, the role of PTEN in WM has not been reported.
IL-6 plays an important role in normal B cell proliferation and maturation and in B-cell malignancies including diffuse large B-cell lymphoma [16], Hodgkin lymphoma [17], and multiple myeloma [18], where it has been shown to regulate the growth of malignant cells. Previous studies have shown that serum IL-6 levels are increased in patients with WM compared to healthy patients [19]. IL-6 has shown a significant upregulation of IgM secretion by WM cells through the CCL5-IL-6-IgM axis in the TME [20][21]. CCL5 signaling has been shown to induce the expression of the transcription factor GLI2 through the PI3K-AKT-IκB-p65 pathway. GLI2 is required to modulate IL-6 expression in vitro and in vivo through this pathway [20]. Targeting the IL-6 receptor with Tocilizumab to block IL-6 effects on WM tumor cells was shown to reduce IgM levels and deter tumor growth in WM, while not inducing toxicity [22]. This suggests that blocking IL-6 may provide therapeutic efficacy in WM. Despite this, targeting IL-6 in WM patients has not been investigated.
The role of bone marrow stromal cells has been extensively studied in WM and are attributed to the growth of WM cells [13][23][24][25]. Ephrin-B2 was demonstrated to be highly expressed on endothelial cells from the bone marrow of patients with WM compared with healthy controls [26] and activation of the Eph-B2 receptor did not directly increase the proliferation of WM cells, but it increased the adhesion of WM cells to endothelial cells, promoting WM cell proliferation [25]. This increase in WM cell proliferation is dependent on downstream activation of focal adhesion kinase (FAK) and Src and inhibition of ephrin-B2 on endothelial cells or inhibition of Eph-B2 on WM cells reduced the adhesion of WM cells to endothelial cells, preventing the proliferative induction from occurring [25].
B-lymphocyte stimulator (BLyS) is a TNF family member expressed by dendritic cells, neutrophils, monocytes, and macrophages and has been shown to be necessary for normal B-cell development. BLyS binds to the receptors B-cell-activating factor of the TNF family receptor (BAFF-R), transmembrane activator and CAML interactor (TACI), and B-cell maturation antigen (BCMA) in WM patients. Expression of BLyS in WM patient bone marrow and elevated serum BLyS levels have also been noted, as well as upregulated IgM secretion upon BLyS addition. In vitro, BLyS was shown to enhance the proliferation and survival of WM cells [27].
Bone marrow mast cells are commonly associated with malignant cells in patients with WM. CD40 ligand (CD40L/CD154) is an inducer of B-cell proliferation and is expressed on malignant cell-associated mast cells in 94% of WM patients, in contrast with 0% of healthy patient mast cell samples. It was found that the co-culture of mast cells and lymphoplasmacytic cells (LPC) induced LPC proliferation and tumor colony formation [28]. Increased Erk phosphorylation and cell growth in malignant B-cells co-cultured with CD40L-expressing stromal cells have also been reported. GLI2 induced increased CD40L expression and GLI2 knockdown decreased CD40L expression. GLI2 has been shown to directly bind to and regulate the activity of the CD40L promoter [29].
2. Survival
Myeloid differentiation factor 88 (MYD88) L265P somatic mutation is present in 91% of WM/LPL patients, per whole genome sequencing results [30][31]. The presence of MYD88 L265P has also been reported in IgM MGUS [32], mucosa-associated lymphoid tissue lymphoma (9%) [33], and diffuse large B-cell lymphoma [34]. Inhibiting MYD88/IRAK signaling induced apoptosis of MYD88 L265P-expressing WM cells by blocking MYD88 homodimerization, an essential process for IRAK1 and IRAK4 signaling (Figure 1). This treatment induced significant apoptosis in BCWM.1, MWCL-1 cell lines as well as primary WM patient cells. Induction of apoptosis did not occur without the MYD88 L265P mutation [30]. Due to the activation of NF-kB, increased anti-apoptotic Bcl-xL expression has been observed in both MYD88 L265P and MYD88 L265RPP mutations, promoting increased survival of malignant cells [35].
3. Angiogenesis
Angiogenesis plays an essential role in wound healing and bone repair and regeneration. This process forms new blood vessels from existing ones, which allow the body to re-establish normal blood flow and oxygen/nutrient/growth factor delivery to the injured or proliferating area [36][37][38][39]. In cancer, tumor cells can develop an angiogenic phenotype through the upregulated pro-angiogenic or downregulated anti-angiogenic pathways [40][41]. This causes endothelial cells to enter a rapid growth phase, forming new blood vessels, and providing nutrients, oxygen, and growth factors to the tumor cells [42]. This process is often rushed in cancer and endothelial cells do not have the time to form perfect blood vessels, leading to leaky, disorganized blood vessels [43][44]. This is an essential step of disease progression and serves to initiate the process of metastasis in many types of cancer [43][45]. VEGF is a well-established growth factor, known for its role in both physiological and pathological angiogenesis. VEGF-A is the main member of the VEGF family and plays a key role in promoting angiogenesis during embryonic development and tissue repair under physiological conditions (Figure 1) [39]. In cancer, VEGF-A production from tumor cells results in an angiogenic switch, leading way to vasculature growth and as a result, tumor growth and metastasis [39]. As the tumor mass increases, the oxygen availability of decreased and hypoxia occurs, leading to the release of proangiogenic factors such as VEGF-A [39]. Angiopoietin-1 (Ang-1) and its antagonist, angiopoietin-2 (Ang-2) serve as the ligands for receptor tyrosine kinase Tie-2 and play a critical role in angiogenesis in both physiological and malignant conditions [46]. Fibroblast growth factors (FGF) are a family of heparin-binding growth factors. Basic FGF (bFGF) interacts with endothelial cell surface receptors and has pro-angiogenic activity [47]. The crosstalk between bFGF, VEGF, and other inflammatory cytokines plays an important role in mediating angiogenesis in the tumor microenvironment.
In WM, the bone marrow microvessel density is only elevated in 30-40% of patients [48]. In a study of 56 patients with WM, it was reported that increased levels of angiogenin, vascular endothelial growth factor (VEGF), vascular endothelial growth factor A (VEGFA), and basic fibroblast growth factor in sera of patients, compared with healthy controls [49]. A lower level of the angiogenesis antagonist, angiopoetin-1 (Ang-1), was also reported in WM sera versus healthy controls [49].
4. Hypoxia
Hypoxia plays an important role in the progression of many malignancies and activated hypoxia pathways are strongly associated with adverse prognosis in cancer [45]. Tumor hypoxia in multiple myeloma activates HIF1α, which promotes cell survival, motility, invasiveness, drug resistance, and neoangiogenesis [50][51] and is associated with a more aggressive tumor [52]. In multiple myeloma, the egress of bone multiple myeloma cells from the bone marrow into the circulation and into new niches was also demonstrated [53].
In a study demonstrating hypoxia in WM cells, the WM cell line, BCWM.1, was genetically engineered to express luciferase and mCherry fluorescent protein. The cells were injected into SCID mice via the tail vein and allowed to grow for 3 weeks to establish tumor burdens in the bone marrow of the mice [52]. This growth in the bone marrow was confirmed by flow cytometry. The mean fluorescent intensity (MFI) of hypoxia marker pimonidazole hydrochloride signal was analyzed and a direct correlation between the tumor burden in the bone marrow and hypoxia in the WM cells was found. Other cells in the bone marrow were tested for hypoxic signs as well and found that the mCherry-negative population was less hypoxic than the WM cells, but still showed hypoxic signs, and hypoxic signs were more greatly shown at higher tumor burdens [52]. In addition, the effect of tumor hypoxia on the egress of WM cells from the bone marrow was tested and a direct linear correlation between the hypoxia in the bone marrow and the number of circulating WM cells was found [52]. This indicated that the mechanism of WM cell entry into circulation is regulated by hypoxia.
Hypoxia also plays a major role in regulating WM cell proliferation. BCWM.1 and MWCL.1 WM cell lines were exposed to normoxic and hypoxic conditions for 24 h in vitro and found that after 24 h of normoxia, the BCWM.1 and MWCL.1 cells had nearly doubled, and the hypoxic cells only increased by 1.3-fold [52]. This suggests that hypoxic conditions do not promote WM cell growth but play a role in other aspects of WM biology.
5. Epithelial–Mesenchymal Transition
The epithelial–mesenchymal transition (EMT) describes a process in which epithelial cells lose their epithelial characteristics and gain a mesenchymal phenotype [54]. This process can lead to increased invasiveness of the cancer cells, leading to overall metastasis [55]. This process allows cancer cells to leave the primary tissue site, enter the bloodstream, and infiltrate other tissues [45].
In a study of WM cells and hypoxia, EMT markers E-cadherin, CXCR4, and VLA-4 were assessed via flow cytometry to determine the effect of hypoxia on EMT in WM. BCWM.1 cells were exposed to either normoxic or hypoxic conditions for 24 h, then analyzed for expression of EMT markers by flow cytometry [52].
The adhesion ability of WM cells to bone marrow stromal cells and to each other was assessed in vitro and incubation of BCWM.1 or MWCL.1 cells in hypoxic conditions reduced their adhesion to a bone marrow stromal cell monolayer by 50% and 25%. This decrease in adhesion was linked to reduced expression of the epithelial marker E-cadherin in WM cells [52].
6. Tumor Spreading and Tissue Infiltration
Ephrin receptors (Eph) represent the largest family of receptor tyrosine kinases (RTK) and are divided into 2 classes: Eph-A and Eph-B, depending on their affinity to ligands ephrin-A and ephrin-B, and they play a critical role in embryogenesis by positioning cells and modulating cell morphology [56][57][58]. As these receptors are not typically found in adult tissue, the presence of EphA1/A2 and ephrin-A1 has been correlated with tumor malignancy and prognosis. The role of these receptors in cancer is still unknown, as they have been found over-expressed in some cancers, but downregulated in others. For example, higher ephrin-A1 expression in liver and colorectal cancer is associated with a worse prognosis [59][60], but in stage I non-small cell lung cancer patients, higher expression levels of EphA2 and ephrin-A1 improved their prognosis [61]. In WM patient samples, the Eph-B2 receptor was found to be overexpressed in WM cells. Inhibition of ephrin-B2 on endothelial cells led to decreased adhesion of WM cells to endothelial cells, therefore decreasing proliferation, cell-cycle progression, and tumor progression in WM cells [26]. In a study looking at the effect of Eph-B2 in WM cells, it was found that inhibition of Eph-B2 on WM cells reduced bone marrow infiltration by WM cells [25].
7. Disease Progression Complications
Bing–Neel syndrome (BNS) is a rare complication of WM. Two types of BNS exist, diffuse and tumoral form. In diffuse form, malignant cells are found in the leptomeningeal space, periventricular white matter, or the spinal cord. The tumoral form is characterized by an intraparenchymal mass or nodular lesion [62]. BNS is rare, with only 1% of patients showing BNS during the disease progression. The treatment of BNS requires drugs with successful infiltration into the central nervous system, such as fludarabine, methotrexate, and cytarabine. Ibrutinib has shown some CNS-penetrating properties and may have a therapeutic role in treating BNS [63].
References
- Hodge, L.S.; Ziesmer, S.C.; Yang, Z.Z.; Secreto, F.J.; Gertz, M.A.; Novak, A.J.; Ansell, S.M. IL-21 in the bone marrow microenvironment contributes to IgM secretion and proliferation of malignant cells in Waldenstrom macroglobulinemia. Blood 2012, 120, 3774–3782.
- Sonvilla, G.; Allerstorfer, S.; Heinzle, C.; Stättner, S.; Karner, J.; Klimpfinger, M.; Wrba, F.; Fischer, H.; Gauglhofer, C.; Spiegl-Kreinecker, S.; et al. Fibroblast growth factor receptor 3-IIIc mediates colorectal cancer growth and migration. Br. J. Cancer 2010, 102, 1145–1156.
- Miyake, M.; Sugano, K.; Sugino, H.; Imai, K.; Matsumoto, E.; Maeda, K.; Fukuzono, S.; Ichikawa, H.; Kawashima, K.; Hirabayashi, K.; et al. Fibroblast growth factor receptor 3 mutation in voided urine is a useful diagnostic marker and significant indicator of tumor recurrence in non-muscle invasive bladder cancer. Cancer Sci. 2009, 101, 250–258.
- Henson, B.; Gollin, S. Overexpression of KLF13 and FGFR3 in Oral Cancer Cells. Cytogenet. Genome Res. 2010, 128, 192–198.
- Van Rhijn, B.W.; Mertens, L.S.; Mayr, R.; Bostrom, P.J.; Real, F.X.; Zwarthoff, E.C.; Boormans, J.L.; Abas, C.; van Leenders, G.J.; Götz, S.; et al. FGFR3 Mutation Status and FGFR3 Expression in a Large Bladder Cancer Cohort Treated by Radical Cystectomy: Implications for Anti-FGFR3 Treatment? Eur. Urol. 2020, 78, 682–687.
- Bersani, C.; Haeggblom, L.; Ursu, R.G.; Giusca, S.E.; Marklund, L.; Ramqvist, T.; Näsman, A.; Dalianis, T. Overexpression of FGFR3 in HPV-positive Tonsillar and Base of Tongue Cancer Is Correlated to Outcome. Anticancer Res. 2018, 38, 4683–4690.
- Long, X.; Shi, Y.; Ye, P.; Guo, J.; Zhou, Q.; Tang, Y. MicroRNA-99a Suppresses Breast Cancer Progression by Targeting FGFR3. Front. Oncol. 2020, 9, 1473.
- Azab, A.K.; Azab, F.; Quang, P.; Maiso, P.; Sacco, A.; Ngo, H.T.; Liu, Y.; Zhang, Y.; Morgan, B.L.; Roccaro, A.M.; et al. FGFR3 Is Overexpressed Waldenström Macroglobulinemia and Its Inhibition by Dovitinib Induces Apoptosis and Overcomes Stroma-Induced Proliferation. Clin. Cancer Res. 2011, 17, 4389–4399.
- O’Donnell, J.S.; Massi, D.; Teng, M.W.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103.
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132.
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203.
- Si, X.; Xu, F.; Xu, F.; Wei, M.; Ge, Y.; Chenge, S. CADM1 inhibits ovarian cancer cell proliferation and migration by potentially regulating the PI3K/Akt/mTOR pathway. Biomed. Pharmacother. 2019, 123, 109717.
- Roccaro, A.M.; Sacco, A.; Husu, E.N.; Pitsillides, C.; Vesole, S.; Azab, A.K.; Azab, F.; Melhem, M.; Ngo, H.T.; Quang, P.; et al. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood 2010, 115, 559–569.
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003, 1, e59.
- Alimonti, A.; Carracedo, A.; Clohessy, J.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458.
- Hashwah, H.; Bertram, K.; Stirm, K.; Stelling, A.; Wu, C.; Kasser, S.; Manz, M.; Theocharides, A.; Tzankov, A.; Müller, A. The IL -6 signaling complex is a critical driver, negative prognostic factor, and therapeutic target in diffuse large B-cell lymphoma. EMBO Mol. Med. 2019, 11, e10576.
- Gholiha, A.R.; Hollander, P.; Glimelius, I.; Hedstrom, G.; Molin, D.; Hjalgrim, H.; Smedby, K.E.; Hashemi, J.; Amini, R.-M.; Enblad, G. Revisiting IL-6 expression in the tumor microenvironment of classical Hodgkin lymphoma. Blood Adv. 2021, 5, 1671–1681.
- Mishra, A.; Dingli, D. Metformin inhibits IL-6 signaling by decreasing IL-6R expression on multiple myeloma cells. Leukemia 2019, 33, 2695–2709.
- Elsawa, S.F.; Ansell, S.M. Cytokines in the Microenvironment of Waldenström’s Macroglobulinemia. Clin. Lymphoma Myeloma 2009, 9, 43–45.
- Elsawa, S.; Almada, L.L.; Ziesmer, S.C.; Novak, A.J.; Witzig, T.E.; Ansell, S.M.; Fernandez-Zapico, M.E. GLI2 Transcription Factor Mediates Cytokine Cross-talk in the Tumor Microenvironment. J. Biol. Chem. 2011, 286, 21524–21534.
- Ansell, S.M.; Grote, D.; Elsawa, S.F.; Gupta, M.; Ziesmer, S.C.; Novak, A.J.; Witzig, T.E. Inhibition of the Jak/Stat Pathway Downregulates Immunoglobulin Production and Induces Cell Death in Waldenström Macroglobulinemia. Blood 2009, 114, 1691.
- Han, W.; Matissek, S.J.; Jackson, D.A.; Sklavanitis, B.; Elsawa, S.F. Targeting IL-6 receptor reduces IgM levels and tumor growth in Waldenström macroglobulinemia. Oncotarget 2019, 10, 3400–3407.
- Ngo, H.T.; Azab, A.K.; Farag, M.; Jia, X.; Melhem, M.M.; Runnels, J.; Roccaro, A.M.; Azab, F.; Sacco, A.; Leleu, X.; et al. Src Tyrosine Kinase Regulates Adhesion and Chemotaxis in Waldenstrom Macroglobulinemia. Clin. Cancer Res. 2009, 15, 6035–6041.
- Leleu, X.; Eeckhoute, J.; Jia, X.; Roccaro, A.M.; Moreau, A.-S.; Farag, M.; Sacco, A.; Ngo, H.T.; Runnels, J.; Melhem, M.R.; et al. Targeting NF-κB in Waldenstrom macroglobulinemia. Blood 2008, 111, 5068–5077.
- Ghobrial, I.M.; Maiso, P.; Azab, A.; Liu, Y.; Zhang, Y.; Issa, G.; Azab, F.; Sacco, A.; Quang, P.; Ngo, H.; et al. The bone marrow microenvironment in Waldenstrom macroglobulinemia. Ther. Adv. Hematol. 2011, 2, 267–272.
- Azab, F.; Azab, A.K.; Maiso, P.; Calimeri, T.; Flores, L.; Liu, Y.; Quang, P.; Roccaro, A.M.; Sacco, A.; Ngo, H.T.; et al. Eph-B2/Ephrin-B2 Interaction Plays a Major Role in the Adhesion and Proliferation of Waldenstrom’s Macroglobulinemia. Clin. Cancer Res. 2012, 18, 91–104.
- Elsawa, S.F.; Novak, A.J.; Grote, D.M.; Ziesmer, S.C.; Witzig, T.E.; Kyle, R.A.; Dillon, S.R.; Harder, B.; Gross, J.A.; Ansell, S.M. B-lymphocyte stimulator (BLyS) stimulates immunoglobulin production and malignant B-cell growth in Waldenström macroglobulinemia. Blood 2006, 107, 2882–2888.
- Tournilhac, O.; Santos, D.D.; Xu, L.; Kutok, J.; Tai, Y.-T.; Le Gouill, S.; Catley, L.; Hunter, Z.; Branagan, A.R.; Boyce, J.A.; et al. Mast cells in Waldenstrom’s macroglobulinemia support lymphoplasmacytic cell growth through CD154/CD40 signaling. Ann. Oncol. 2006, 17, 1275–1282.
- Ghobrial, I.M.; Roccaro, A.; Hong, F.; Weller, E.; Rubin, N.; Leduc, R.; Rourke, M.; Chuma, S.; Sacco, A.; Jia, X.; et al. Clinical and Translational Studies of a Phase II Trial of the Novel Oral Akt Inhibitor Perifosine in Relapsed or Relapsed/Refractory Waldenström’s Macroglobulinemia. Clin. Cancer Res. 2010, 16, 1033–1041.
- Han, W.; Jackson, D.; Matissek, S.J.; Misurelli, J.A.; Neil, M.S.; Sklavanitis, B.; Amarsaikhan, N.; Elsawa, S.F. Novel Molecular Mechanism of Regulation of CD40 Ligand by the Transcription Factor GLI2. J. Immunol. 2017, 198, 4481–4489.
- Yang, G.; Zhou, Y.; Liu, X.; Xu, L.; Cao, Y.; Manning, R.J.; Patterson, C.J.; Buhrlage, S.J.; Gray, N.; Tai, Y.-T.; et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013, 122, 1222–1232.
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833.
- Xu, L.; Hunter, Z.; Yang, G.; Zhou, Y.; Cao, Y.; Liu, X.; Morra, E.; Trojani, A.; Greco, A.; Arcaini, L.; et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013, 121, 2051–2058.
- Gachard, N.; Parrens, M.; Soubeyran, P.-L.; Petit, B.; Marfak, A.; Rizzo, D.; Devesa, M.; Delage-Corre, M.; Coste, V.; Laforêt, M.P.; et al. IGHV gene features and MYD88 L265P mutation separate the three marginal zone lymphoma entities and Waldenström macroglobulinemia/lymphoplasmacytic lymphomas. Leukemia 2012, 27, 183–189.
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837.
- Nagao, T.; Oshikawa, G.; Ishida, S.; Akiyama, H.; Umezawa, Y.; Nogami, A.; Kurosu, T.; Miura, O. A novel MYD88 mutation, L265RPP, in Waldenström macroglobulinemia activates the NF-κB pathway to upregulate Bcl-xL expression and enhances cell survival. Blood Cancer J. 2015, 5, e314.
- Rizov, M.; Andreeva, P.; Dimova, I. Molecular regulation and role of angiogenesis in reproduction. Taiwan. J. Obstet. Gynecol. 2017, 56, 127–132.
- Augustine, R.; Prasad, P.; Khalaf, I.M.N. Therapeutic angiogenesis: From conventional approaches to recent nanotechnology-based interventions. Mater. Sci. Eng. C 2019, 97, 994–1008.
- Zimta, A.-A.; Baru, O.; Badea, M.; Buduru, S.D.; Berindan-Neagoe, I. The Role of Angiogenesis and Pro-Angiogenic Exosomes in Regenerative Dentistry. Int. J. Mol. Sci. 2019, 20, 406.
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.-L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 295–342.
- Guerra, A.; Belinha, J.; Jorge, R.N. Modelling skin wound healing angiogenesis: A review. J. Theor. Biol. 2018, 459, 1–17.
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370.
- Dalal, B.; Quinn, T.J.; Foster, L.; Lin, M.; Matthews, M.; Yuhan, B. Ligand-directed tumor targeting with hybrid viral phage nanoparticles. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 483–516.
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; de la Cruz, O.N.H.; Lopez-Gonzalez, J.S. Contribution of Angiogenesis to Inflammation and Cancer. Front. Oncol. 2019, 9, 1399.
- Boutilier, A.; Elsawa, S. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995.
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248.
- Yu, X.; Ye, F. Role of Angiopoietins in Development of Cancer and Neoplasia Associated with Viral Infection. Cells 2020, 9, 457.
- Wang, J.; Li, L.; Jiang, M.; Li, Y. MicroRNA-195 inhibits human gastric cancer by directly targeting basic fibroblast growth factor. Clin. Transl. Oncol. 2017, 19, 1320–1328.
- Terpos, E.; Tasidou, A.; Kastritis, E.; Eleftherakis-Papaiakovou, E.; Gavriatopoulou, M.; Migkou, M.; Dimopoulos, M.-A. Angiogenesis in Waldenström’s Macroglobulinemia. Clin. Lymphoma Myeloma 2009, 9, 46–49.
- Anagnostopoulos, A.; Zervas, K.; Kastritis, E.; Tsionos, K.; Bamias, A.; Meletis, J.; Dimopoulos, M.; Terpos, E.; Eleftherakis-Papaiakovou, V. Serum concentrations of angiogenic cytokines in Waldenstrom macroglobulinaemia: The ratio of angiopoietin-1 to angiopoietin-2 and angiogenin correlate with disease severity. Br. J. Haematol. 2007, 137, 560–568.
- Storti, P.; Bolzoni, M.; Donofrio, G.; Airoldi, I.; Guasco, D.; Toscani, D.; Martella, E.; Lazzaretti, M.; Mancini, C.; Agnelli, L.; et al. Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia 2013, 27, 1697–1706.
- Zhang, J.; Sattler, M.; Tonon, G.; Grabher, C.; Lababidi, S.; Zimmerhackl, A.; Raab, M.S.; Vallet, S.; Zhou, Y.; Cartron, M.-A.; et al. Targeting Angiogenesis via a c-Myc/Hypoxia-Inducible Factor-1α–Dependent Pathway in Multiple Myeloma. Cancer Res. 2009, 69, 5082–5090.
- Muz, B.; de la Puente, P.; Azab, F.; Ghobrial, I.M.; Azab, A.K. Hypoxia Promotes Dissemination and Colonization in New Bone Marrow Niches in Waldenström Macroglobulinemia. Mol. Cancer Res. 2015, 13, 263–272.
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794.
- Diepenbruck, M.; Christofori, G. Epithelial–mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13.
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52.
- Lodola, A.; Giorgio, C.; Incerti, M.; Zanotti, I.; Tognolini, M. Targeting Eph/ephrin system in cancer therapy. Eur. J. Med. Chem. 2017, 142, 152–162.
- Niethamer, T.K.; Bush, J.O. Getting direction(s): The Eph/ephrin signaling system in cell positioning. Dev. Biol. 2019, 447, 42–57.
- Wada, H.; Yamamoto, H.; Kim, C.; Uemura, M.; Akita, H.; Tomimaru, Y.; Hama, N.; Kawamoto, K.; Kobayashi, S.; Eguchi, H.; et al. Association between ephrin-A1 mRNA expression and poor prognosis after hepatectomy to treat hepatocellular carcinoma. Int. J. Oncol. 2014, 45, 1051–1058.
- Yamamoto, H.; Tei, M.; Uemura, M.; Takemasa, I.; Uemura, Y.; Murata, K.; Fukunaga, M.; Ohue, M.; Ohnishi, T.; Ikeda, K.; et al. Ephrin-A1 mRNA is associated with poor prognosis of colorectal cancer. Int. J. Oncol. 2012, 42, 549–555.
- Simon, L.; Fitsiori, A.; Lemal, R.; Dupuis, J.; Carpentier, B.; Boudin, L.; Corby, A.; Aurran-Schleinitz, T.; Gastaud, L.; Talbot, A.; et al. Bing-Neel syndrome, a rare complication of Waldenstrom macroglobulinemia: Analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015, 100, 1587–1594.
- Castillo, J.J.; Treon, S.P. How we manage Bing–Neel syndrome. Br. J. Haematol. 2019, 187, 277–285.
- Ishikawa, M.; Miyahara, R.; Sonobe, M.; Horiuchi, M.; Mennju, T.; Nakayama, E.; Kobayashi, M.; Kikuchi, R.; Kitamura, J.; Imamura, N.; et al. Higher expression of EphA2 and ephrin-A1 is related to favorable clinicopathological features in pathological stage I non-small cell lung carcinoma. Lung Cancer 2012, 76, 431–438.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
717
Revisions:
2 times
(View History)
Update Date:
08 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No