Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mark Tingey | -- | 5037 | 2022-10-05 22:32:53 | | | |
| 2 | Lindsay Dong | -9 word(s) | 5028 | 2022-10-08 07:15:49 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tingey, M.; Schnell, S.J.; Yu, W.; Saredy, J.; Junod, S.; Patel, D.; Alkurdi, A.A.; Yang, W. Technologies Enabling Single-Molecule Super-Resolution Imaging of mRNA. Encyclopedia. Available online: https://encyclopedia.pub/entry/28273 (accessed on 24 June 2026).
Tingey M, Schnell SJ, Yu W, Saredy J, Junod S, Patel D, et al. Technologies Enabling Single-Molecule Super-Resolution Imaging of mRNA. Encyclopedia. Available at: https://encyclopedia.pub/entry/28273. Accessed June 24, 2026.
Tingey, Mark, Steven J. Schnell, Wenlan Yu, Jason Saredy, Samuel Junod, Dhrumil Patel, Abdullah A. Alkurdi, Weidong Yang. "Technologies Enabling Single-Molecule Super-Resolution Imaging of mRNA" Encyclopedia, https://encyclopedia.pub/entry/28273 (accessed June 24, 2026).
Tingey, M., Schnell, S.J., Yu, W., Saredy, J., Junod, S., Patel, D., Alkurdi, A.A., & Yang, W. (2022, October 05). Technologies Enabling Single-Molecule Super-Resolution Imaging of mRNA. In Encyclopedia. https://encyclopedia.pub/entry/28273
Tingey, Mark, et al. "Technologies Enabling Single-Molecule Super-Resolution Imaging of mRNA." Encyclopedia. Web. 05 October, 2022.
Copy Citation
The transient nature of RNA has rendered it one of the more difficult biological targets for imaging. This difficulty stems both from the physical properties of RNA as well as the temporal constraints associated therewith. These concerns are further complicated by the difficulty in imaging endogenous RNA within a cell that has been transfected with a target sequence. These concerns, combined with traditional concerns associated with super-resolution light microscopy has made the imaging of this critical target difficult.
mRNA
single-molecule super-resolution microscopy
FISH
smFISH
Aptamer
seqFISH
SPEED microscopy
s9.6 antibody
MTRIPS
Molecular Beacons
MERFISH
MCP-MS2 loop system
SMLM
dCAS13
CRISPR
dCAS9
1. Introduction
1.1. Image RNA
The flow of genetic information is a multi-stage process centered around RNA metabolism. Since Francis Crick proposed the central dogma of molecular biology [1], RNA has been a central focus in the field of molecular and cellular biology. RNA is a multi-functional macromolecule that plays an essential role in gene expression and regulation. In gene expression, messenger RNA (mRNA) acts as templates that carry the genetic information from the DNA blueprint to ribosomes. Transfer RNA (tRNA) acts as an amino acid transporter that helps decode an mRNA sequence into a protein. Ribosomal RNA (rRNA), one of the main components of the ribosome, has shown deep involvement in ribosomal subunit association, tRNA binding, and translocation during translation [2][3]. In gene regulation, an overwhelming amount of evidence has demonstrated that small regulatory RNA is associated with cellular regulation via various mechanisms.
1.2. RNA Localization and Imaging: Seeing Is Believing
Controlling the localization of RNA is a widespread, evolutionarily conserved, and efficient way to target gene products to a specific region of a cell or embryo [4]. Using mRNA as an example, after the initial transcription, mRNA proceeds through post-transcriptional modification such as alternative splice, nucleocytoplasmic transportation across the nuclear pore complex, localization to the ribosome, translation, and finally degradation. These steps are highly coordinated and tightly regulated both spatially and temporally. Unsurprisingly, the subcellular localization of RNA has been determined to be one of the fundamental mechanisms of cell polarization. One good example is β-actin mRNA expression in moving fibroblast cells, where mRNAs for β-actin are concentrated at the moving edge.
In order to obtain a complete spatial-temporal profile of RNA throughout its entire lifespan, from transcription to degradation, methods to visualize RNA within cells are required. Such methods are critically important to enhance the understanding of RNA and thus offer unparalleled opportunities for advancement in cellular and molecular biology, therapeutic discovery, and medical diagnostic.
1.3. Difficulties Associated with Imaging mRNA
Generally speaking, the size of mRNA is far smaller than the resolution limit of conventional light microscopy. Thus, only two approaches are feasible regarding mRNA imaging: electron microscopy (EM) and fluorescent light microscopy. Among various EM techniques, two techniques have been used in mRNA imaging: first, an EM-level adaptation of in situ hybridization (ISH) technique that combines antisense probes and gold-coupled antibodies for detection [5][6]; and second, Cryo-EM, a mainstream technology in structural biology for architectural study [7][8]. The EM approach for mRNA imaging provides excellent spatial resolution, structural, and ultrastructural information. However, it has limitations, including the lack of temporal resolution due to the use of fixed samples, overly complicated sample preparation, artifact susceptibility [9], and low labeling efficiency [10]. EM-ISH is still a commonly used technique for obtaining images with a high spatial resolution that reveal the cellular distribution of mRNA in fixed tissue, whereas Cryo-EM is the predominant method for mRNA structure. Observation of mRNA in live tissue is required to explore mRNA transport dynamics, maintenance and regulatory mechanisms, and localization.
An appropriate fluorophore used to label RNA needs to have three outstanding characteristics: quantum yield, extinction coefficient [11], and photostability. The relative brightness of a fluorophore is dependent upon the quantum yield and extinction coefficient of the fluorophore, causing some fluorophores to emit more light than others under the same excitation power. Thus, it can provide better spatial and temporal resolution without risking cell viability in live cell imaging. At the same time, fluorophore stability is associated with its resistance to photobleaching. Photobleaching is a dynamic process in which a fluorophore exposed to excitation light undergoes photoinduced chemical destruction, thus losing its ability to fluoresce [12]. A fluorophore with higher stability will stay unphotobleached for an extended time, allowing for more prolonged imaging.
In the past two decades, a series of revolutionary techniques termed super-resolution microscopy (SRM) has been developed that bypass the diffraction limit. The diffraction limit is a barrier in optical microscopy caused by the physical property of light, restricting the optical resolution to roughly 250 nm [13]. There are generally two subgroups of SRM [14], the first being SRM by single-molecule-localization-based imaging such as stochastic optical reconstruction microscopy (STORM) [15] and photoactivated localization microscopy (PALM) [16] and the second being SRM by spatially patterned excitation such as stimulated emission depletion (STED) microscopy [17] and structured illumination microscopy (SIM) [18]. STED and SIM achieve super-resolution using patterned illumination to differentially modulate the fluorescence emission of molecules within the diffraction-limited volume [19]. Thus, those techniques have been well used with fixed samples labeled with ISH and immunofluorescence to obtain sub-diffraction images [20][21][22]. On the other hand, SRM by single-molecule-localization-based imaging, enables the determination of the localization of a single fluorophore. Currently, research involving trajectory and single molecule tracking of mRNA is primarily performed using single-molecule localization microscopy (SMLM) techniques in live samples [23][24][25].
1.4. Single-Molecule Super-Resolution Imaging
SMLM methods typically utilize conventional wide-field excitation and achieve super-resolution by fitting and localizing individual molecules, which are subsequently utilized to form a complete image in a pointillistic fashion [25]. Since the inception of these techniques, they have become broadly adopted in life science research because of their superior spatial resolution, which in most cases can achieve 20 nm lateral and 50 nm axial resolution or better [13]. SMLM is fundamentally based on the fact that the spatial coordinates of single fluorescent molecules can be determined with high precision from an isolated point spread function (PSF). The single PSF can be approximated with a Gaussian intensity distribution, allowing the exact center of the corresponding single emitter to be determined, even if it sits between two pixels of the imaging system [26]. In order to avoid overlapping between PSFs, fluorescent emissions of distinct molecules have to be separate. There are other ways to ensure a temporal separation between PSFs; the most commonly used approach exploits photoswitchable or photoactivation probes. Supposing the majority of fluorophores in a sample are converted to a dark state, and only a tiny subset of the population switches back on, the probability of two emitters residing near each other will be minimal. Under these conditions, one can calculate and record the location of each emitter in this subset. After bleaching or switching off the current emissive fluorophores, a new subset can be activated. This process can be repeated multiple times. When a sufficient number of location data are accumulated, the structure associated with the fluorophores can be reconstructed from hundreds of subsets of emitter distribution.
1.5. Utilizing Single-Molecule Super-Resolution Imaging for mRNA
Before diving into various SMLM techniques used to image mRNA, the advantages and disadvantages of live cell imaging and fixed samples must first be addressed. Fluorescence microscopy of living or fixed cells is entirely dependent upon suitable labeling and detection strategies. In fixed cells, because all cellular activities and movements have been terminated during fixation, this imaging strategy allows repeat capture to maximize localization precisions, which contributes to the spatial resolution advantages of STORM and PALM. However, cell fixation also has significant limitations.
Live cell imaging is a closer representation to the natural state of the cell. Whereas fixed cells are best described as in situ, live cells remain the best method of obtaining data regarding the behavior of cells in their native environments. For example, live cell imaging enables researchers to obtain real-time measurements at the temporal frequency necessary to sample the dynamics of most biological processes adequately [27].
Another consideration when comparing the two primary super-resolution approaches is the size of the dataset required to generate super-resolution localizations. Depending on the samples and applications involved, SMLM methodologies often require thousands of frames to reconstruct a high-quality image from an individual localization event, sometimes requiring minutes or hours to capture. As a result, many SMLM techniques are often performed on chemically fixed cells to prevent cell movement and facilitate the localization of single molecules. Despite this consideration, SMLM techniques have enabled significant progress in the arena of imaging mRNA dynamics and mRNA-protein interactions.
RNA has also been studied utilizing the single-particle tracking capacity of SMLM methodologies. The principles of SMLM can be expanded to track single-particles within live cells where a labeled single RNA macromolecule is localized with nanometer precision and observed over a period of time. Such fine-detailed and dynamic information, when paired with structural information of the cell, provides invaluable information pertaining to the dynamics and transport behaviors of RNA. A distinct advantage of tracking single particles is the ability to derive 3D information regarding the dynamic behaviors of RNA. One way in which this has been utilized is through the use of single-particle localization in two-dimensions to derive virtual three-dimensional (3D) information using a computational algorithm via single-point edge-excitation sub-diffraction (SPEED) microscopy. SPEED microscopy has been used to track messenger ribonucleoprotein (mRNP) movement through the nuclear pore complex (NPC) of eukaryotic cells [23][28][29]. This technique is specifically designed to track and record 2D spatial locations of fast-moving fluorophores within a rotationally symmetric biological structure with a spatiotemporal resolution of approximately 10 nm and 0.4 ms, respectively [23][28][29]. After image acquisition, post-localization 2D-to-3D transformation is applied to obtain 3D super-resolution structural and dynamic information. Besides the computational 2D-to-3D algorithm specially designed for rotational symmetric biological channels in the nuclear pore, another SMLM approach combined with multifocus microscopy (MFM) was also developed within the past few years. The MFM method produces focal stacks of high-resolution 2D images simultaneously displayed on a single camera [30].
2. RNA Imaging Methods
2.1. FISH
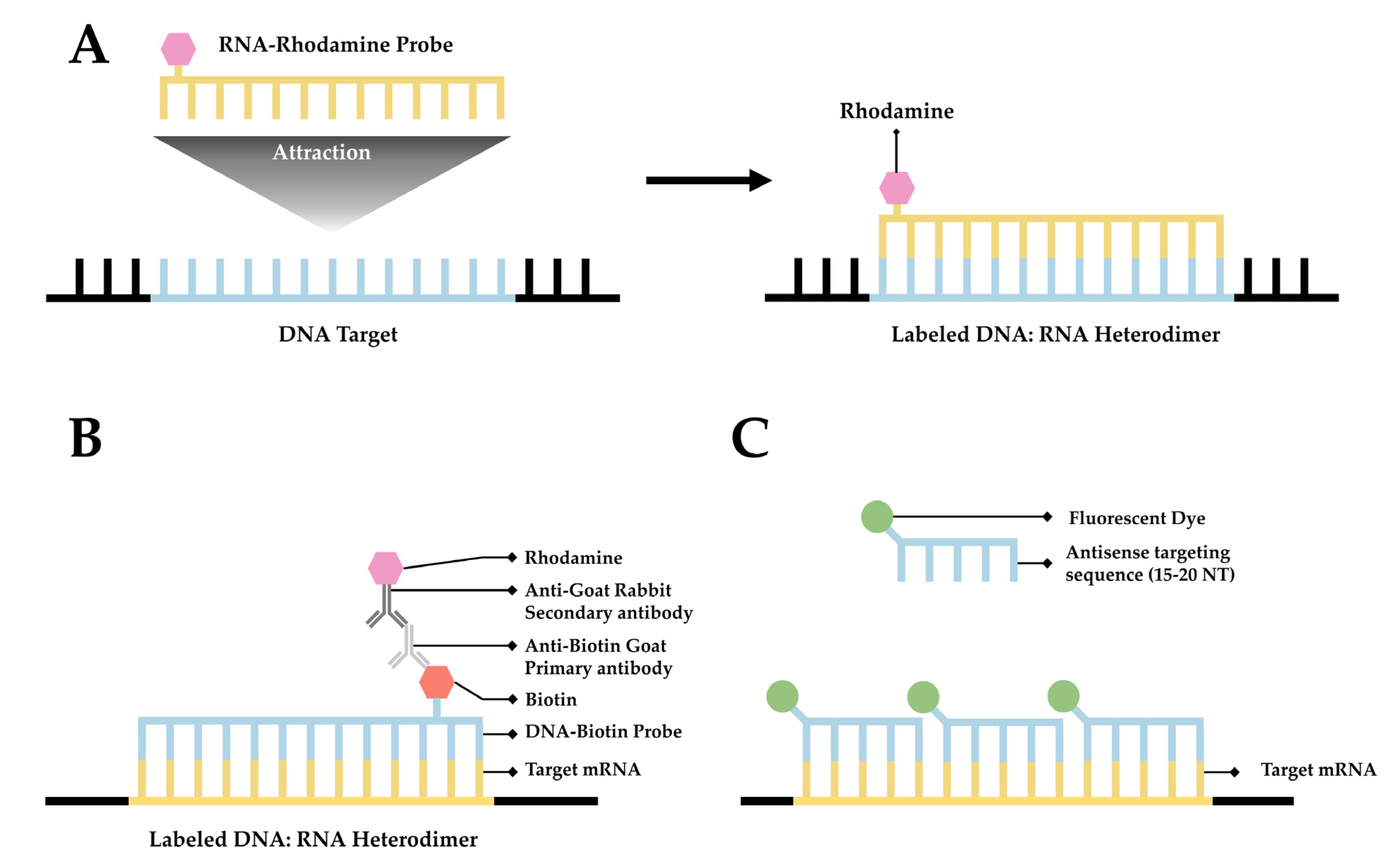
Fluorescence in situ hybridization (FISH) is a valuable method for imaging nucleic acids. First developed by Bauman and colleagues in 1980 [31], this method was originally intended to be a way to replace and improve on old methods of in situ hybridization (ISH) that utilized 3H- or 125I-labeled radioactive hybridization probes. By combining the hybridization approach in the previous autoradiography methods with observations made by Rudkin & Stollar [32], wherein it was demonstrated that RNA:DNA hybrids could be targeted via a fluorescently labeled antibody, Bauman and colleagues devised a mechanism to specifically target a nucleic acid by introducing a complimentary RNA sequence covalently bound to a fluorescent probe that would then hybridize with the desired sequence [31] (Figure 1A).
The early incarnation of FISH was shown to facilitate imaging of mitochondrial DNA, viral DNA within human tissue culture cells, and 5S rRNA [31]. Whereas this technique demonstrated the theoretical possibility of FISH to image hybridized RNA, the first true mRNA FISH was performed by Singer & Ward in 1982 to visualize actin mRNA in chicken skeletal muscle culture [33]. This visualization was accomplished using a DNA probe labeled with biotin. After the hybridization of DNA to RNA, a goat anti-biotin primary antibody followed by a rabbit derived anti-goat secondary antibody conjugated to rhodamine was added [33] (Figure 1B). This complex showed higher fluorescence than the direct comparison approach originally devised by Bauman and colleagues [34]; however, the size of the complex made it unwieldy.
In response to these difficulties, ISH methodology has been improved over the intervening years, primarily focusing on two areas: the development of new and novel fluorophores and the addition of multiple fluorophores to a single probe. The original incarnation of FISH used a single fluorophore, TRITC (tetra-methyl rhodamine isothiocyanate), to label RNA [31]. Likewise, Singer and Ward utilized a single Rhodamine conjugated to an antibody in the first iteration of RNA FISH [33]. Later studies employed multiple probes with differing fluorophores to interrogate the relationships and localizations of multiple target DNA sequences simultaneously. This approach resulted in the development of multiplex-FISH (M-FISH), a technique in which multiple targets are simultaneously tagged with up to 24 different fluorophores, enabling clinicians to screen karyotypes for deleterious genetic mutations [35][36]. This system was eventually adapted to RNA with the development of sequential-FISH (seqFISH).
One of the significant problems with mRNA FISH remains the photostability of the fluorophores used, particularly when attempting super-resolution, as the photon flux required in super-resolution environments results in the rapid deterioration of fluorophores during excitation [37][38]. One response to this problem has been to move away from organic fluorophores. An intriguing recent advance in this arena has been the development of quantum dots, which exhibit brightness equal to organic dyes with significantly improved photostability.
The second approach to improving upon ISH methodology, the addition of multiple fluorophores to a single probe, was first utilized in RNA FISH by attaching multiple CY3 fluorophores to a DNA probe. This approach resulted in significantly higher fluorescence and enabled researchers to identify discrete sequences of mRNA enabling the quantification of mRNA for the first time via mRNA FISH. Although this advance is often considered the first incarnation of single-molecule FISH (smFISH) [39], this approach faced significant technical challenges, as the close proximity of the CY3 fluorophores resulted in self-quenching [34][40]. This resulted in differential fluorescence intensity of individual probes, making quantification via this methodology unreliable. Although this particular iteration was unable to resolve single particles, the idea of multiple fluorophores generating an additive fluorescent intensity was a good one and eventually resulted in the development of smFISH in 2008 [38].
Figure 1. A simplified diagram depicting fluorescent in situ hybridization (FISH). (A) The first FISH experiment in DNA performed by Bauman and colleagues [31], in which a target DNA sequence (Blue) forms and RNA:DNA heterodimer with an RNA (Green) probe conjugated with Rhodamine (Red). (B) The first mRNA FISH experiment performed by Singer & Ward [33], in which a target mRNA sequence (Green) formed a DNA:RNA heterodimer with a complimentary DNA sequence (Blue) conjugated to biotin (purple). A primary anti-Biotin Goat derived primary antibody (Dark Blue) associates with the biotin tag. A secondary anti-Goat rabbit derived antibody (Light Red) conjugated to a Rhodamine (Dark Red) then associates with the primary antibody forming a complete fluorescent label. (C) The core principle of single-molecule FISH (smFISH), in which a target mRNA (Green) is targeted with short sequential antisense oligonucleotides (Blue), each 15–20 nucleotides long, that are each conjugated to a fluorescent dye (Dark Green).
2.2. smFISH
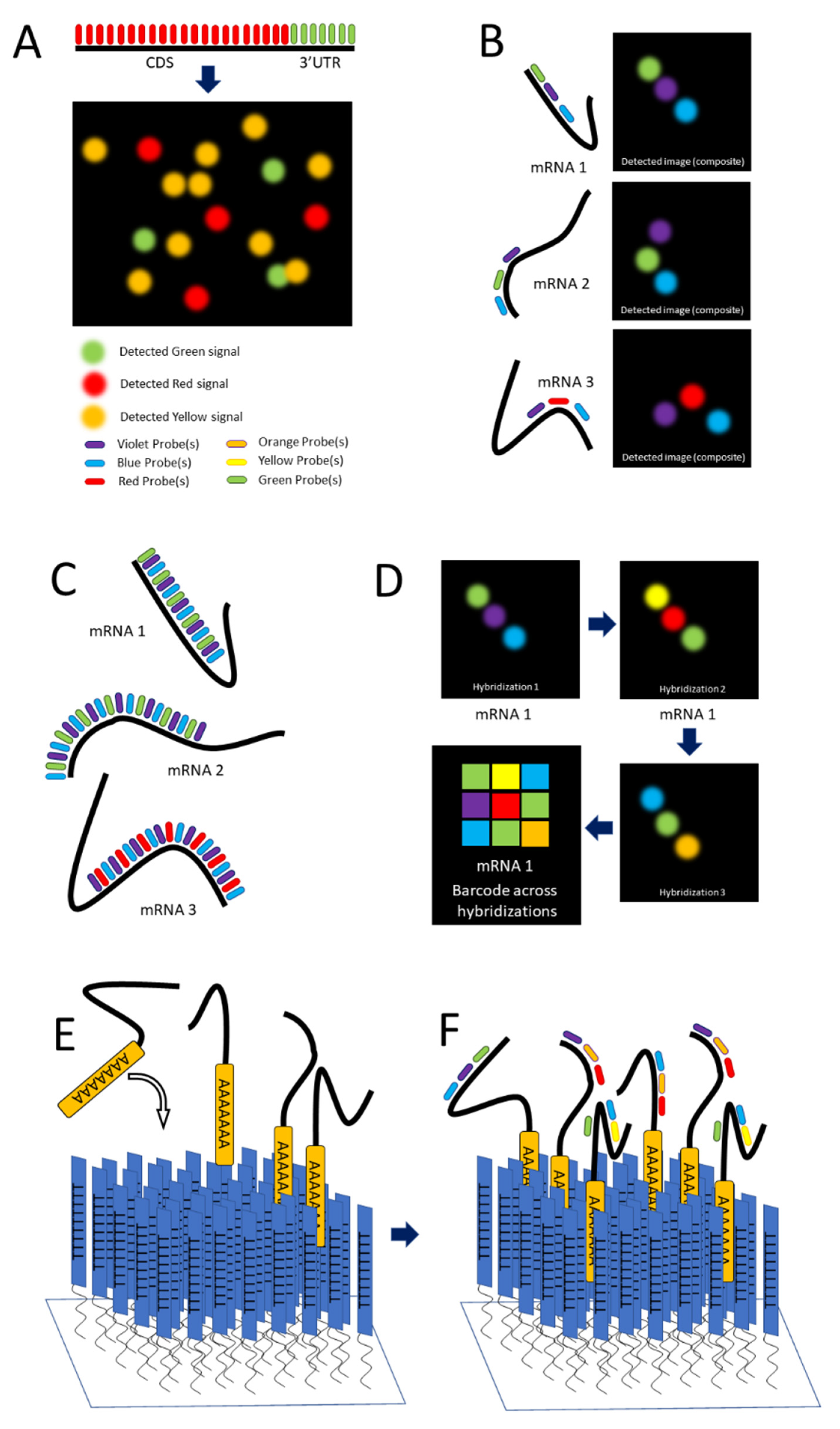
Single-molecule FISH (smFISH) was developed by Raj and colleagues in 2008 to address limitations in FISH concerning low intensity of signal [38]. The scholars tackled this problem by producing a series of 15 to 20 nucleotide long antisense oligonucleotide (ASO) probes, each conjugated to a fluorescent dye (Figure 1C and Figure 2A), along the length of the coding DNA sequence (CDS) and 3′ untranslated region (UTR) regions of intended mRNA transcript. The additive intensity of these multiple probes enabled easier detection of the transcripts such that they could be imaged, localized, and counted (Figure 2A) [38]. Using different combinations of spectrally distinct probes, multiple mRNAs can be imaged simultaneously; however, because of potential spectral overlap, the number of possible combinations is small (See Figure 2A). (The scholars demonstrate three distinct mRNAs).
While smFISH has many technical strengths and has been invaluable in answering many questions regarding mRNA behavior within the cell, it suffers from many significant shortcomings. This technique is viable only in fixed cells. This presents four significant problems to imaging mRNA. First, static information derived from a fixed cells only provides researchers with a snapshot of their behavior. Second, the fixation of a cell often alters membrane structures, potentially leading investigators to draw potentially erroneous conclusions regarding interactions between mRNA and the nuclear envelope [41]. Third, the assay is technically challenging to perform and requires assay optimization for each target. This is due to the presence of too many ASOs producing untenable background noise. Alternatively, if too few ASOs are present, the data cannot be trusted as the full transcript is unlikely to be labeled. Finally, smFISH relies upon qRT-PCR for relative quantification. This further complicates this already technically demanding method with the addition of another technically demanding technique.
2.3. seqFISH
Using techniques such as smFISH to resolve and identify mRNAs is limited by their proximity, which is frequently below the optical diffraction limit. This potentially causes spectral overlap and the loss of discrete signal. Whereas temporal separation of detection is a key feature of other single-molecule approaches such as SPEED microscopy [29][42][43][44][45], it requires a low concentration of tagged molecules in the field of detection. For techniques such as SPEED, which is particularly suited to the dynamic environments of live cells, the need to keep the concentration of detected molecules low presents a limitation. In techniques that are more suitable for static environments, such as STORM [15][46] and STED [47], the need for low concentration of probes can be overcome to some degree with the use of multiple colors of fluorophores [48] if fixed cells are used.
Lubeck and Cai in 2012 and Shah and colleagues in 2016 described sequential FISH (seqFISH) as a way to overcome these limitations to result in a technique that resolved transcripts from 32 stress-responsive genes in single S. cerevisiae cells by combining spatial and spectral coding using (spatial) order of probes (along a transcript) and combinations of colors [49][50]. Their first approach was combinatorial labeling, in which activator-emitter probes were spaced along the mRNA far enough apart (about 100 nucleotides) to be resolved at the resolution capable with STORM. (The localization resolution of STORM is approximately 20 nm [15].) The scholars hybridized probes of varying spectral patterns in particular order to different mRNAs to differentiate them, terming this technique spatial barcoding [49] (see Figure 2B).
Figure 2. smFISH and seqFISH. (A) smFISH: Top, multiple probes of the same color designed to hybridize along the length of both the CDS and 3′UTR of the intended transcript; bottom, mRNAs imaged after hybridization, with yellow spots showing transcripts containing both the CDS and 3′UTR [after (Raj, 2008)]. (B) seqFISH Spatial barcoding, in which probes are designed to hybridize ~100 nt apart to facilitate resolution of unique combinations (after [49]). (C) Spectral barcoding, in which a color code of probes hybridizes repeatedly along the length of a transcript in order to increase its detectability and identifiability. (D) Repeated cycles of hybridization, imaging, and removal of probes results in a temporal barcode that increases the number of unique barcodes possible and aids resolution via the temporal dimension (Panels C,D after [51]). © seqFISH in vitro: An oligo(dT) surface is created and mRNAs hybridize to it via their poly-A tail, spreading out to a resolvable distance. (F) Probes are hybridized to the adhered transcripts.
2.4. MERFISH
It is currently desirable to obtain information regarding the transcriptome localization during various cellular states. To this end, multiplexed error-robust FISH (MERFISH) was developed by Chen and colleagues in 2015 [52]. MERFISH relies upon similar methodologies as seqFISH and smFISH, where multiple antisense oligomers are utilized similar to smFISH. The difference between smFISH and MERFISH becomes apparent as this technique adds each fluorescently tagged antisense oligormer in a sequential manner, thereby uniquely labeling RNA in a manner similar to seqFISH. Where seqFISH creates a color barcode, MERFISH relies upon a 16-bit coding approach.
This technique provides researchers with a robust, error-resistant methodology for quantifying and localizing specific RNA of interest within the cell. Similar to smFISH, this method can only be utilized in fixed cells as the immobility of the RNA is critical to the success of this method. This makes this an ideal technique for capturing whole transcriptome information at key points in the cell life cycle, but not well-suited for capturing details regarding RNA dynamics. Further, producing so many smFISH probes is costly and untenable; to address this, the scholars have adapted an existing Oligopaint approach [53][54]. This approach also requires a degree of coding competence to be able to both generate the ‘codebook’ of target RNA sequences and automate the localization and association of the individual sequences with their designated code word. Lastly, this approach may be hampered by possible photodamage. Theoretically, this technique can be expanded to cover the entirety of the transcriptome; however, the photobleaching required between each hybridization could potentially damage the target cell after multiple sequential rounds of hybridization and imaging.
2.5. Single-Molecule Localization Microscopy (SMLM)
SMLM is a broad discipline that covers a wide array of methods and techniques that have variously been utilized to image RNA. All of these techniques fundamentally rely upon the principles of spatial localization, meaning that sufficient temporal or physical space is required between individual fluorophores to permit the mathematical fitting of the point-spread function (PSF) without overlapping, thereby determining the 2D localization as well as the localization precision [25][29][55]. The separation of individual PSFs can be accomplished utilizing specialized fluorophores such as photoswitching or photoactivated fluorophores for STORM and PALM, respectively.
3. RNA Labeling Strategies
3.1. MCP-MS2 Loop System
The MCP-MS2 loop system is the current gold standard for labeling mRNA in living cells and has been utilized for numerous studies in a variety of model systems over the past few decades [23][28][56][57][58][59][60][61][62][63]. This technique is derived from the virus Emesvirus zinderi also known as Bacteriophage MS2 (MS2), a virus that stands out for being the first genome ever fully sequenced [64][65]. MS2 is a single positive-strand genomic RNA virus that infects Gram-negative bacteria with a retractile pilus and contains a genome of ~4 kb that encodes for four proteins: the maturation protein, the coat protein, the replicase, and the lysis protein [66][67]. Following entry via pilus, the virion goes through uncoating, exposing its genome. The MS2 genome is then cleaved, translated, and replicated. Much of the MS2 genome produces stem-loop structures following transcription that prevent translation. During assembly, the coat MS2 coat protein (MCP) recognizes these stem-loop RNA structures, binds to them, and facilitates encapsidation [68].
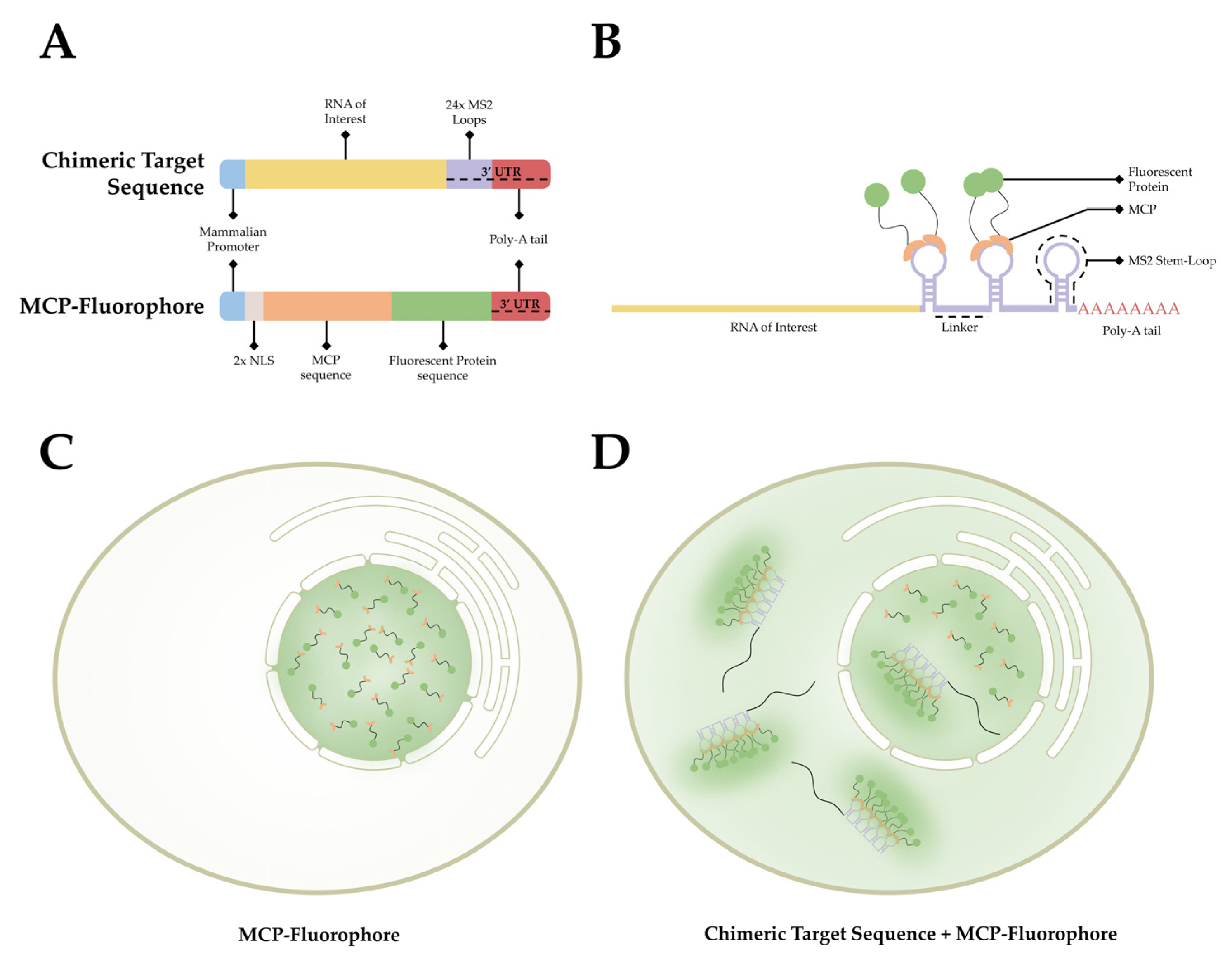
This observed process has provided researchers with a method to label RNA in living organisms. As stem-loop structures do not exist in mammalian cells, MCP does not interact with macromolecules endogenous to mammalian cells. This affords researchers the ability to develop a bipartite labeling methodology, where two plasmids would be introduced to a cell of interest. First, a plasmid containing a mammalian promoter, the sequence encoding for the RNA of interest, and the MS2 loop sequences in the 3′ UTR. Second, a plasmid containing a mammalian promoter, nuclear localization signals (NLS), and the sequence for MCP conjugated with a fluorophore on the 3′ end (Figure 3A). When both plasmids are present and transcribed/translated, an RNA sequence containing MS2 loops at the 3′ end is present, as well as a nuclear-localized MCP-fluorophore. The target sequence and the MCP-Fluorophore then bind with high affinity, creating an MCP-MS2 array that can be localized using light microscopy (Figure 3B). It is important to note that the addition of the NLS to the MCP-Fluorophore enables researchers to ensure that both plasmids are present in the cell, as cells in which fluorescence is only present within the nucleus were lacking the target RNA sequence. This is due to the fact that MCP-Fluorophore binding to mRNA will be exported to the cytoplasm, thereby causing fluorescence, however dim, to be present in the cytoplasm as well as the nucleus (Figure 3C,D).
Figure 3. The MCP-MS2 loop system. (A) A depiction of the gene cassettes present in the two plasmids, the chimeric target sequence and the MCP-fluorophore, utilized in this system. (B) A simplified diagram of the association between the chimeric target sequence and the MCP-Fluorophore post transcription/translation. (C) A depiction of the fluorescent pattern observed in cells that have only the MCP-Fluorophore plasmid. (D) A depiction of the fluorescent pattern observed in cells that contain both the MCP-Fluorophore and Chimeric Target Sequence plasmids.
In summation, the MCP-MS loop system provides several distinct advantages detailed here, most notably being the ease of use in live cells. However, there are also many distinct disadvantages to this system. Chief among the disadvantages is the high background noise present in the system, as unbound MCP-fluorophore conjugates aggregate in the nucleus. Next, the MS2 loop, as it is not present in mammalian cells, confounds cellular machinery. Chimeric mRNA transcripts are untranslatable, and the tight bonding between the MCP-MS2 loop impairs accessibility of mRNA decay enzymes to the MS2 array, leading to slow degradation in S. cerevisiae [69][70][71][72]. This deficiency presents significant difficulty for researchers interrogating the full life cycle and intracellular localization of mRNA. The issue has been addressed by developing new versions of the MS2 loop system, specifically by adjusting the linker space between the individual stem-loops. This adjustment is hypothesized to create more space to allow endogenous proteins to interact with the chimeric sequence, thereby reducing or potentially ameliorating this impairment [73][74].
3.2. Antibody Labeling
Several steps within the life cycle of an mRNA molecule may inhibit or prematurely end its progression to the ribosome. One of these inhibitory instances, occurring during transcription, is the formation of an RNA:DNA hybrid, R-Loop, from single-stranded RNA hybridization with the complementary DNA sequence. Numerous factors may cause RNA:DNA hybrid formation, relaxed upstream supercoiling, defective proteins related to stalled transcription and RNA:DNA hybrid resolution, G-rich mRNA regions, and non-template DNA strand nicks and secondary structures [75].
3.3. Multiply-Labeled Tetravalent RNA Imaging Probes (MTRIPS)
Imaging endogenous mRNA is preferred as it provides a more accurate picture of biological processes unaffected by plasmid overexpression or other experimental artifacts arising from transfection. Further, endogenous mRNA also avoids restriction to cell types that can be efficiently transfected. Studying mRNA expression in live cells can provide dynamic information about how mRNA expression changes in response to varying conditions or over time.
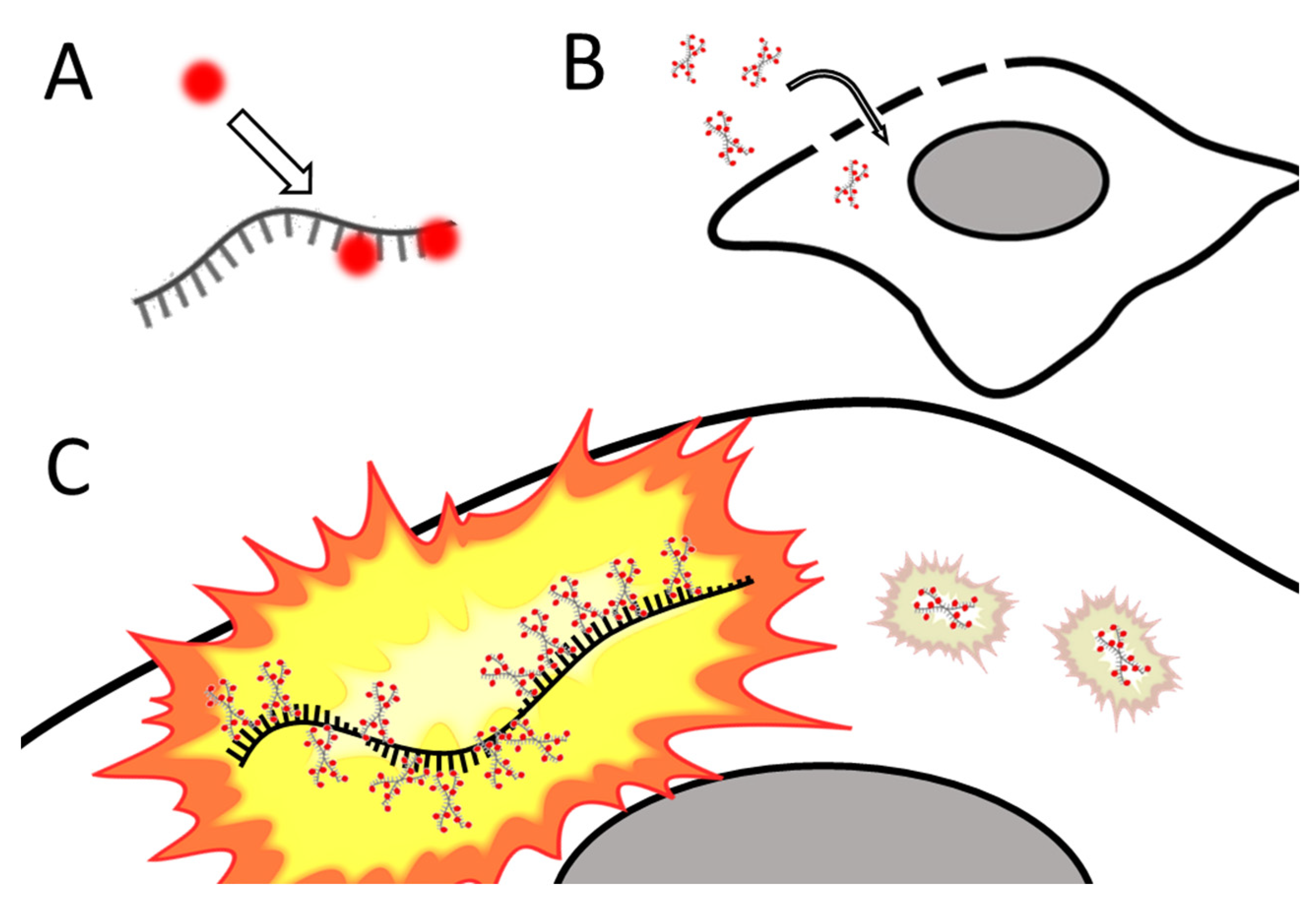
Santangelo and colleagues developed multiply labeled tetravalent RNA imaging probes (MTRIPs), a method of labeling native mRNA transcripts with multiple fluorophores [76]. Synthetic oligos (2′ O-methyl RNA-DNA chimera nucleic acid ligands) were labeled with multiple fluorophores, bound to streptavidin, and delivered into the live cell via reversible permeabilization with streptolysin O. After entry into the cytosol, multiple ligands bind to target mRNA transcripts (see Figure 4). Unbound probes and mRNAs bound to few probes can be eliminated by measuring differential intensity; points of light showing the intensity level expected for one probe are disregarded; mRNAs bound to multiple probes show a higher signal-to-noise ratio. The scholars leveraged the technique they developed to image RNA at the single-molecule level, showing colocalization of RNA with RNA-binding proteins in live human epithelial cancer cells and primary chicken fibroblasts [76]. Later innovation resulted in the development of proximity ligation assays utilizing MTRIPS [77], thereby providing more versatility to this labeling strategy.
Figure 4. Simplified schematic of MTRIPs. (A) Fluorophores are bound to synthetic oligomers (ligands). (B) Tagged ligands are introduced into the live cell via temporary permeabilization. (C) Ligands bind to target mRNA (not o scale); multiply bound mRNAs are distinguished from unbound probes by intensity. After [76].
3.4. CRISPR-Based Labeling Strategies
Using endogenous tagging of mRNA such as the previously mentioned MS2 loop systems would be further improved by the ability to shorten the endogenous tag insertion steps and flexibility in insertion sites. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is a hot topic field in genomic manipulation, first discovered in 1987 [78] and pushed into development as a tool for genetic modifications concurrently by Charpentier [79] and Zhang [80]. Using a sequence-specific targeting Single guide RNA (sgRNA), it can precisely target excision sites to insert exogenous tags into th’ 3’ UTR of mRNA sequences [81][82]. Many online tools have been developed to help find appropriate sgRNAs and protospacer sequences to target virtually any gene: “Zhang Lab.” Available online: http://crispr.mit.edu (accessed on 8 August 2022), “Benchling.” Available online: http://benchling.com (accessed on 8 August 2022). These tools are most effectively used in conjunction with Zhang’s protocol for assembling gRNA plasmids [83].
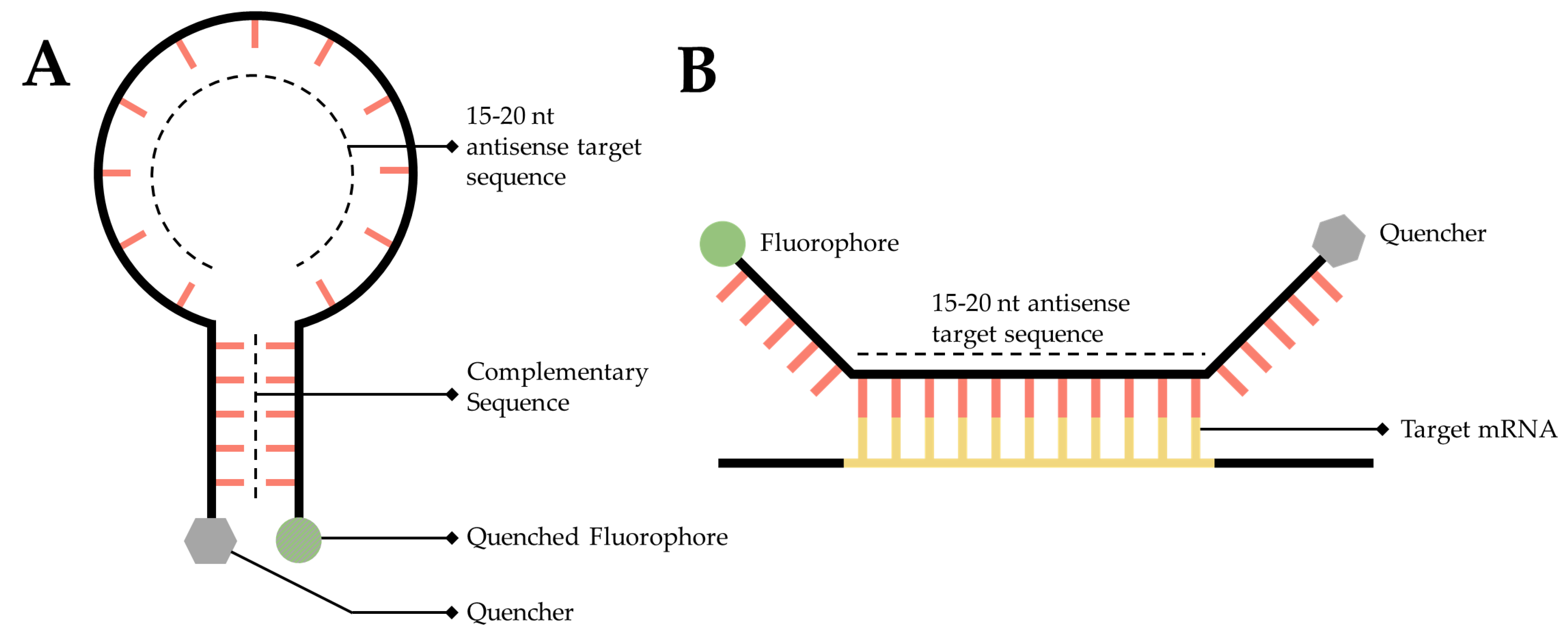
3.5. RNA Molecular Beacons
An RNA targeted molecular beacon functions along a similar premise as all ISH methodologies but is especially similar to smFISH. Specifically, a short, 15- to 20-nt, fluorescently tagged DNA oligo binds to a complimentary transcript of interest within the cell, thereby enabling researchers to visualize and localize that transcript. The primary difference is that this technology attempts to resolve the background noise problem inherent to so many of the mRNA labeling techniques discussed in this manuscript. Researchers have accomplished this by adding a fluorophore to one end of the sequence and a quencher to the other [84] (Figure 5A). A quencher is a compound that, when sufficiently close to a fluorophore, will absorb the energy released by the fluorophore and dissipate it as heat [84][85]. The quencher is utilized to great effect by designing the beacon as a stem-loop, flanking the 15- to 20-nt antisense target sequence. When the beacon is not bound to its target, the beacon forms a hairpin, bringing the fluorophore and quencher into close proximity (Figure 5A), thereby generating a probe that will only fluoresce when it is bound to the target (Figure 5B) [86].
Figure 5. Simplified diagram of RNA molecular beacons. (A) A 15- to 20-nucleotide target sequence flanked by palindromic repeats causes the probe to form a stem-loop, bringing the quencher (Grey) and a fluorophore into close proximity causing the fluorophore to quench (Dark Grey). (B) When in close proximity to the target transcript, the target sequence will hybridize with the target mRNA, causing the stem-loop to open, moving the quencher and fluorophore away from one another, thereby facilitating fluorescence.
3.6. RNA Aptamer
An aptamer is a single-stranded length of DNA or RNA that forms a secondary structure that selectively binds to a specific target [87]. As there are no known fluorescent RNAs, this is an attractive feature for imaging RNA. Conditional fluorophore aptamers, also known as fluorescent turn-on aptamers, have been developed to enable the imaging of RNA. These aptamers form a specific secondary structure which will then selectively bind to a specific dye that exhibits minimal fluorescence until it binds with its cognate aptamer. This is made possible as a result of the following three principles, twisted intramolecular charge transfer, excited state proton transfer, and unquenching of fluorophore-quencher conjugates [88]. Each of these results in a physical change to the dye enabling it to emit fluorescence upon excitation when associated with the aptamer.
This system has been utilized in a variety of studies to evaluate the localization and behavior of telomerase-associated RNA in both mammalian cells and Saccharomyces cerevisiae [89][90][91][92]; specifically, TLC1 [93], the RNA scaffold for the telomerase holoenzyme, and telomeric repeat-containing RNA (TERRA), a DNA:RNA heteroduplex that actively participates in the telomere maintenance and chromosome end protection [94]. Providing valuable information regarding the localization and dynamics of these RNAs within both dividing and senescent cells.
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563.
- Noller, H.F. Ribosomal RNA and translation. Annu. Rev. Biochem. 1991, 60, 191–227.
- Elela, S.A.; Nazar, R.N. Role of the 5.8 S rRNA in ribosome translocation. Nucleic Acids Res. 1997, 25, 1788–1794.
- Kloc, M.; Zearfoss, N.R.; Etkin, L.D. Mechanisms of subcellular mRNA localization. Cell 2002, 108, 533–544.
- Delanoue, R.; Herpers, B.; Soetaert, J.; Davis, I.; Rabouille, C. Drosophila Squid/hnRNP helps Dynein switch from a gurken mRNA transport motor to an ultrastructural static anchor in sponge bodies. Dev. Cell 2007, 13, 523–538.
- Herpers, B.; Rabouille, C. mRNA localization and ER-based protein sorting mechanisms dictate the use of transitional endoplasmic reticulum-golgi units involved in gurken transport in Drosophila oocytes. Mol. Biol. Cell 2004, 15, 5306–5317.
- Galej, W.P.; Toor, N.; Newman, A.J.; Nagai, K. Molecular mechanism and evolution of nuclear pre-mRNA and group II intron splicing: Insights from cryo-electron microscopy structures. Chem. Rev. 2018, 118, 4156–4176.
- Wilkinson, M.E.; Lin, P.-C.; Plaschka, C.; Nagai, K. Cryo-EM studies of pre-mRNA splicing: From sample preparation to model visualization. Annu. Rev. Biophys. 2018, 47, 175–199.
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158.
- Weil, T.T.; Parton, R.M.; Davis, I. Making the message clear: Visualizing mRNA localization. Trends Cell Biol. 2010, 20, 380–390.
- Weber, G.; Teale, F. Determination of the absolute quantum yield of fluorescent solutions. Trans. Faraday Soc. 1957, 53, 646–655.
- Song, L.; Hennink, E.; Young, I.T.; Tanke, H.J. Photobleaching kinetics of fluorescein in quantitative fluorescence microscopy. Biophys. J. 1995, 68, 2588–2600.
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72–84.
- Huang, B.; Bates, M.; Zhuang, X. Super resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993.
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796.
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645.
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782.
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87.
- Sigal, Y.M.; Zhou, R.; Zhuang, X. Visualizing and discovering cellular structures with super-resolution microscopy. Science 2018, 361, 880–887.
- Somasekharan, S.P.; Saxena, N.; Zhang, F.; Beraldi, E.; Huang, J.N.; Gentle, C.; Fazli, L.; Thi, M.; Sorensen, P.H.; Gleave, M. Regulation of AR mRNA translation in response to acute AR pathway inhibition. Nucleic Acids Res. 2022, 50, 1069–1091.
- Harbauer, A.B.; Hees, J.T.; Wanderoy, S.; Segura, I.; Gibbs, W.; Cheng, Y.; Ordonez, M.; Cai, Z.; Cartoni, R.; Ashrafi, G. Neuronal mitochondria transport Pink1 mRNA via Synaptojanin 2 to support local mitophagy. Neuron 2022, 110, 1516–1531.e1519.
- Zhang, W.I.; Röhse, H.; Rizzoli, S.O.; Opazo, F. Fluorescent in situ hybridization of synaptic proteins imaged with super-resolution STED microscopy. Microsc. Res. Tech. 2014, 77, 517–527.
- Li, Y.; Aksenova, V.; Tingey, M.; Yu, J.; Ma, P.; Arnaoutov, A.; Chen, S.; Dasso, M.; Yang, W. Distinct roles of nuclear basket proteins in directing the passage of mRNA through the nuclear pore. Proc. Natl. Acad. Sci. USA 2021, 118.
- Paramasivam, P.; Franke, C.; Stöter, M.; Höijer, A.; Bartesaghi, S.; Sabirsh, A.; Lindfors, L.; Arteta, M.Y.; Dahlén, A.; Bak, A. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 2021, 221, e202110137.
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffié, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Primers 2021, 1, 1–27.
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 2018, 6, 022003.
- Sung, M.H.; McNally, J.G. Live cell imaging and systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 167–182.
- Ma, J.; Liu, Z.; Michelotti, N.; Pitchiaya, S.; Veerapaneni, R.; Androsavich, J.R.; Walter, N.G.; Yang, W. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat. Commun. 2013, 4, 1–9.
- Li, Y.; Tingey, M.; Ruba, A.; Yang, W. High-speed super-resolution imaging of rotationally symmetric structures using SPEED microscopy and 2D-to-3D transformation. Nat. Protoc. 2021, 16, 532–560.
- Abrahamsson, S.; Chen, J.; Hajj, B.; Stallinga, S.; Katsov, A.Y.; Wisniewski, J.; Mizuguchi, G.; Soule, P.; Mueller, F.; Darzacq, C.D. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat. Methods 2013, 10, 60–63.
- Bauman, J.; Wiegant, J.; Borst, P.; Van Duijn, P. A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochrome-labelled RNA. Exp. Cell Res. 1980, 128, 485–490.
- Rudkin, G.T.; Stollar, B. High resolution detection of DNA–RNA hybrids in situ by indirect immunofluorescence. Nature 1977, 265, 472–473.
- Singer, R.H.; Ward, D.C. Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc. Natl. Acad. Sci. USA 1982, 79, 7331–7335.
- Young, A.P.; Jackson, D.J.; Wyeth, R.C. A technical review and guide to RNA fluorescence in situ hybridization. PeerJ 2020, 8, e8806.
- Kearney, L. Multiplex-FISH (M-FISH): Technique, developments and applications. Cytogenet. Genome Res. 2006, 114, 189–198.
- Speicher, M.R.; Ballard, S.G.; Ward, D.C. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat. Genet. 1996, 12, 368–375.
- Liu, Y.; Le, P.; Lim, S.J.; Ma, L.; Sarkar, S.; Han, Z.; Murphy, S.J.; Kosari, F.; Vasmatzis, G.; Cheville, J.C. Enhanced mRNA FISH with compact quantum dots. Nat. Commun. 2018, 9, 1–8.
- Raj, A.; Van Den Bogaard, P.; Rifkin, S.A.; Van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879.
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590.
- Randolph, J.B.; Waggoner, A.S. Stability, specificity and fluorescence brightness of multiply-labeled fluorescent DNA probes. Nucleic Acids Res. 1997, 25, 2923–2929.
- Tingey, M.; Mudumbi, K.C.; Schirmer, E.C.; Yang, W. Casting a Wider Net: Differentiating between Inner Nuclear Envelope and Outer Nuclear Envelope Transmembrane Proteins. Int. J. Mol. Sci. 2019, 20, 5248.
- Ma, J.; Yang, W. Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots. Proc. Natl. Acad. Sci. USA 2010, 107, 7305–7310.
- Ruba, A.; Kelich, J.; Ma, J.; Yang, W. Reply to ‘Deconstructing transport-distribution reconstruction in the nuclear-pore complex’. Nat. Struct. Mol. Biol. 2018, 25, 1062–1064.
- Schnell, S.J.; Tingey, M.; Yang, W. Speed Microscopy: High-Speed Single Molecule Tracking and Mapping of Nucleocytoplasmic Transport. In The Nuclear Pore Complex: Methods and Protocols; Goldberg, M.W., Ed.; Springer: New York, NY, USA, 2022; pp. 353–371.
- Schnell, S.J.; Ma, J.; Yang, W. Three-dimensional mapping of mRNA export through the nuclear pore complex. Genes 2014, 5, 1032–1049.
- Bates, M.; Dempsey, G.T.; Chen, K.H.; Zhuang, X. Multicolor super-resolution fluorescence imaging via multi-parameter fluorophore detection. ChemPhysChem 2012, 13, 99–107.
- Westphal, V.; Hell, S.W. Nanoscale resolution in the focal plane of an optical microscope. Phys. Rev. Lett. 2005, 94, 143903.
- Miklyaev, Y.V.; Asselborn, S.; Zaytsev, K.; Darscht, M.Y. Superresolution microscopy in far-field by near-field optical random mapping nanoscopy. Appl. Phys. Lett. 2014, 105, 113103.
- Lubeck, E.; Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 2012, 9, 743–748.
- Shah, S.; Lubeck, E.; Zhou, W.; Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 2016, 92, 342–357.
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361.
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090.
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.; Li, J.B.; Senaratne, T.N.; Williams, B.R. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306.
- Moffitt, J.R.; Zhuang, X. RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). Methods Enzymol 2016, 572, 1–49.
- von Diezmann, L.; Shechtman, Y.; Moerner, W. Three-dimensional localization of single molecules for super-resolution imaging and single-particle tracking. Chem. Rev. 2017, 117, 7244–7275.
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell 1998, 2, 437–445.
- Grünwald, D.; Singer, R.H. In vivo imaging of labelled endogenous β-actin mRNA during nucleocytoplasmic transport. Nature 2010, 467, 604–607.
- Larson, D.R.; Zenklusen, D.; Wu, B.; Chao, J.A.; Singer, R.H. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 2011, 332, 475–478.
- Wu, B.; Eliscovich, C.; Yoon, Y.J.; Singer, R.H. Translation dynamics of single mRNAs in live cells and neurons. Science 2016, 352, 1430–1435.
- Halstead, J.M.; Lionnet, T.; Wilbertz, J.H.; Wippich, F.; Ephrussi, A.; Singer, R.H.; Chao, J.A. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 2015, 347, 1367–1671.
- Morisaki, T.; Lyon, K.; DeLuca, K.F.; DeLuca, J.G.; English, B.P.; Zhang, Z.; Lavis, L.D.; Grimm, J.B.; Viswanathan, S.; Looger, L.L. Real-time quantification of single RNA translation dynamics in living cells. Science 2016, 352, 1425–1429.
- Lim, B.; Levine, M.; Yamazaki, Y. Transcriptional pre-patterning of Drosophila gastrulation. Curr. Biol. 2017, 27, 286–290.
- Tantale, K.; Mueller, F.; Kozulic-Pirher, A.; Lesne, A.; Victor, J.-M.; Robert, M.-C.; Capozi, S.; Chouaib, R.; Bäcker, V.; Mateos-Langerak, J. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat. Commun. 2016, 7, 1–14.
- Paranchych, W. Attachment, ejection and penetration stages of the RNA phage infectious process. RNA Phages 1975, 85–111.
- Fiers, W.; Contreras, R.; Duerinck, F.; Haegeman, G.; Iserentant, D.; Merregaert, J.; Min Jou, W.; Molemans, F.; Raeymaekers, A.; Van den Berghe, A. Complete nucleotide sequence of bacteriophage MS2 RNA: Primary and secondary structure of the replicase gene. Nature 1976, 260, 500–507.
- Zinder, N.D. Portraits of Viruses: RNA Phage. Intervirology 1980, 13, 257–270.
- Gorzelnik, K.V.; Zhang, J. Cryo-EM reveals infection steps of single-stranded RNA bacteriophages. Prog. Biophys Mol. Biol. 2021, 160, 79–86.
- Rolfsson, Ó.; Middleton, S.; Manfield, I.W.; White, S.J.; Fan, B.; Vaughan, R.; Ranson, N.A.; Dykeman, E.; Twarock, R.; Ford, J. Direct evidence for packaging signal-mediated assembly of bacteriophage MS2. J. Mol. Biol. 2016, 428, 431–448.
- Garcia, J.F.; Parker, R. MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: Implications for the localization of mRNAs by MS2-MCP system. RNA 2015, 21, 1393–1395.
- Garcia, J.F.; Parker, R. Ubiquitous accumulation of 3′ mRNA decay fragments in Saccharomyces cerevisiae mRNAs with chromosomally integrated MS2 arrays. RNA 2016, 22, 657–659.
- Haimovich, G.; Zabezhinsky, D.; Haas, B.; Slobodin, B.; Purushothaman, P.; Fan, L.; Levin, J.Z.; Nusbaum, C.; Gerst, J.E. Use of the MS2 aptamer and coat protein for RNA localization in yeast: A response to “MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: Implications for the localization of mRNAs by MS2-MCP system”. RNA 2016, 22, 660–666.
- Heinrich, S.; Sidler, C.L.; Azzalin, C.M.; Weis, K. Stem–loop RNA labeling can affect nuclear and cytoplasmic mRNA processing. RNA 2017, 23, 134–141.
- Vera, M.; Tutucci, E.; Singer, R.H. Imaging Single mRNA Molecules in Mammalian Cells Using an Optimized MS2-MCP System. In Imaging Gene Expression: Methods and Protocols; Shav-Tal, Y., Ed.; Springer New York: New York, NY, USA, 2019; pp. 3–20.
- Tutucci, E.; Vera, M.; Biswas, J.; Garcia, J.; Parker, R.; Singer, R.H. An improved MS2 system for accurate reporting of the mRNA life cycle. Nat. Methods 2018, 15, 81–89.
- Chedin, F.; Benham, C.J. Emerging roles for R-loop structures in the management of topological stress. J. Biol. Chem. 2020, 295, 4684–4695.
- Santangelo, P.J.; Lifland, A.W.; Curt, P.; Sasaki, Y.; Bassell, G.J.; Lindquist, M.E.; Crowe, J.E. Single molecule–sensitive probes for imaging RNA in live cells. Nat. Methods 2009, 6, 347–349.
- Jung, J.; Lifland, A.W.; Zurla, C.; Alonas, E.J.; Santangelo, P.J. Quantifying RNA–protein interactions in situ using modified-MTRIPs and proximity ligation. Nucleic Acids Res. 2012, 41, e12.
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433.
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.L.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.B.; Jiang, W.Y.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823.
- Spille, J.H.; Hecht, M.; Grube, V.; Cho, W.K.; Lee, C.; Cissé, I.I. A CRISPR/Cas9 platform for MS2-labelling of single mRNA in live stem cells. Methods 2019, 153, 35–45.
- Wang, M.; Chen, K.; Wu, Q.; Peng, R.; Zhang, R.; Li, J. RCasFISH: CRISPR/dCas9-Mediated in Situ Imaging of mRNA Transcripts in Fixed Cells and Tissues. Anal. Chem. 2020, 92, 2468–2475.
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308.
- Tyagi, S.; Kramer, F.R. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996, 14, 303–308.
- George, L.; Indig, F.E.; Abdelmohsen, K.; Gorospe, M. Intracellular RNA-tracking methods. Open Biol. 2018, 8.
- Li, Y.; Zhou, X.; Ye, D. Molecular beacons: An optimal multifunctional biological probe. Biochem. Biophys. Res. Commun. 2008, 373, 457–461.
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550.
- Trachman, R.J.; Ferré-D’Amaré, A.R. Tracking RNA with light: Selection, structure, and design of fluorescence turn-on RNA aptamers. Q. Rev. Biophys. 2019, 52, e8.
- Sato, H.; Das, S.; Singer, R.H.; Vera, M. Imaging of DNA and RNA in Living Eukaryotic Cells to Reveal Spatiotemporal Dynamics of Gene Expression. Annu. Rev. Biochem. 2020, 89, 159–187.
- Li, X.; Liu, L.; Yin, F.; Liu, Y.; Xu, S.; Jiang, W.; Wang, R.; Xue, Q. In situ generation spatial confinement fluorescence RNA for sensitive and stable imaging of telomerase RNA in cells. Sens. Actuators B Chem. 2021, 345, 130414.
- Laprade, H.; Lalonde, M.; Guérit, D.; Chartrand, P. Live-cell imaging of budding yeast telomerase RNA and TERRA. Methods 2017, 114, 46–53.
- Tingey, M. Yeast Npl3 Regulates TERRA Production and Prevents Faster Senescence in Telomerase-Null Cells. Master’s Thesis, Saint Joseph’s University, Francis A. Drexel Library, Philadelphia, PA, USA, 2017.
- Zappulla, D.C. Yeast Telomerase RNA Flexibly Scaffolds Protein Subunits: Results and Repercussions. Molecules 2020, 25, 2750.
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246.
More
Information
Subjects:
Biophysics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
08 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No