Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Siqiniseko Sinikiwe Ndlovu | -- | 1923 | 2022-09-30 21:13:30 | | | |
| 2 | Sirius Huang | Meta information modification | 1923 | 2022-10-08 03:53:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ndlovu, S.S.; Ghazi, T.; Chuturgoon, A.A. Toxic Effects of Highly Active Antiretroviral Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/28189 (accessed on 26 July 2026).
Ndlovu SS, Ghazi T, Chuturgoon AA. Toxic Effects of Highly Active Antiretroviral Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/28189. Accessed July 26, 2026.
Ndlovu, Siqiniseko S., Terisha Ghazi, Anil A. Chuturgoon. "Toxic Effects of Highly Active Antiretroviral Therapy" Encyclopedia, https://encyclopedia.pub/entry/28189 (accessed July 26, 2026).
Ndlovu, S.S., Ghazi, T., & Chuturgoon, A.A. (2022, September 30). Toxic Effects of Highly Active Antiretroviral Therapy. In Encyclopedia. https://encyclopedia.pub/entry/28189
Ndlovu, Siqiniseko S., et al. "Toxic Effects of Highly Active Antiretroviral Therapy." Encyclopedia. Web. 30 September, 2022.
Copy Citation
Highly active antiretroviral therapy (HAART) comprises a combination of two or three antiretroviral (ARV) drugs that are administered together in a single tablet. These drugs target different steps within the human immunodeficiency virus (HIV) life cycle, providing either a synergistic or additive antiviral effect; this enhances the efficiency in which viral replication is suppressed. HIV cannot be completely eliminated, making HAART a lifetime treatment. With long-term HAART usage, an increasing number of patients experience a broadening array of complications, and this significantly affects their quality of life, despite cautious use.
HIV
HAART
pathophysiology
metabolic syndrome
Moringa oliefera
1. Introduction
The World Health Organization (WHO) reported that there are 38 million people currently living with human immunodeficiency virus (HIV) globally, and the majority of these individuals are in South Africa (SA) [1]. SA has an HIV infection prevalence of 19% and carries the largest disease burden worldwide [2][3]. HIV is suppressed through the effective use of antiretroviral (ARV) drugs [4]. Over the past years, ARV formulations have been improved, and when combined with two or three ARVs from different ARV drug classes, make a highly active antiretroviral therapy (HAART), also known as antiretroviral therapy (ART).
The implementation of HAART prolongs the life expectancy in HIV-infected individuals, and HAART has led to a significant decline in morbidity and mortality among HIV-infected patients [5][6]. Despite its high effectiveness to suppress HIV viral replication, HAART cannot completely eliminate the virus because of the presence of multiple T-cell reservoirs [7] and, for this reason, HIV-infected patients need to be on HAART throughout their lifetime in order to keep their viral load under 50 copies/mL [8][9]. As a result of HAART being a life-long treatment, adverse outcomes associated with this long-term therapy have been emerging.
HAART has evolved with the intention to make it less toxic, while optimizing its function; however, it is not void of toxicity. The ART regimen of tenofovir disoproxil fumarate (TDF), lamivudine (3TC), emtricitabine (FTC), dolutegravir (DTG), and efavirenz (EFV), in the long-term, has been associated with the development of pathophysiological complications, referred to as metabolic syndrome (MetS) [10][11][12]. MetS is a combination of metabolic disorders that include hypertension, hyperglycemia, changes in fat distribution, increased cholesterol low-density lipoprotein (LDL) and triglycerides, and reduced levels of cholesterol high-density lipoprotein (HDL), which may lead to cardiovascular diseases (CVDs) such as heart disease, stroke, and diabetes [13][14][15][16][17][18][19]. It is therefore of paramount importance that constant approaches for the improvement of ART treatment are made a clinical and pharmaceutical priority. Alternatively, the use of supplementary medicine such as medicinal/herbal plants may provide a possible solution.
2. HAART-Induced Mitochondrial Toxicity and Oxidative Stress
The long-term use of ARV drugs contributes to long-term complications in HIV-infected persons. Mitochondrial dysfunction and oxidative stress are highlighted as metabolic pathways through which ARV drugs induce MetS [20][21][22][23]. Nucleoside reverse transcriptase inhibitors (NRTIs), which are a cornerstone of HAART regimens, non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors, and integrase strand transfer inhibitors (PIs/INSTIs) have been noticeably associated with many adverse effects related to mitochondrial toxicity and oxidative stress [24][25][26][27].
The effects of HAART were observed in the mitochondria (Figure 1). NRTIs found in HAART inhibit the activity of DNA polymerase-γ, an enzyme responsible for the replication and maintenance of mitochondrial DNA (mtDNA), thus compromising mitochondrial integrity and function [23][28]. The triphosphate (active) forms of NRTI are potential substrates for the polymerase-γ and can provoke the termination of the DNA chain during mtDNA replication [29]. The mtDNA depletion also leads to an impaired synthesis of mtDNA encoded respiratory chain polypeptides, which can partially block the flow of electrons in the respiratory chain. As a result, they accumulate in complex I and III, where they react with oxygen to form the superoxide anion radical [30]. These effects have been described with selected NRTIs [31][32][33].

Figure 1. An overview of long-term HAART mitochondrial toxicity and oxidative stress in HIV-positive individuals. HAART interferes with the synthesis of polymerase-γ, reducing the mtDNA. This therapy impairs ETC, increasing ROS production, depolarizing the mitochondrial membrane, and compromising the ATP synthesis. HAART also depletes GSH and other cellular antioxidants, propagating oxidative stress in the cell. Created with BioRender.com (access date: 2 June 2022).
The NRTIs impairs oxidative phosphorylation (OXPHOS) proteins and increases oxidative stress in the mitochondria. This leads to damage of mitochondrial proteins and lipids further impairing mitochondrial function [34][35] Mitochondrial dysfunction by NRTIs is also manifested by depolarization of the mitochondrial membrane and increased reactive oxygen species (ROS) generation [32]. NRTI further interferes with the synthesis of essential proteins of the mitochondrial electron transport chain (ETC), causing alterations in nucleotide phosphorylation, directly interfering with mitochondrial respiration and reduce ATP production [36][37]. NRTIs also impair respiration and ATP synthesis, by preventing ATP/ADP translocation [37].
Not all NRTIs exert the same degree of polymerase-γ inhibition; however, they have the capacity to induce mitochondrial toxicity. In vitro studies have demonstrated that 3TC inhibits polymerase-γ, although its affinity for polymerase-γ is not as strong as the previously discontinued ARVs [38][39]. Samuels, Bayerri [40] reported that mtDNA deletion mutation was detectable significantly more commonly in the urine of TDF exposed study participants as compared to unexposed individuals.
FTC effects on the mitochondria include reduction of ATP synthesis and mitochondrial membrane depolarisation [33]. FTC has also been shown to cause mitochondrial dysfunction when used together with TDF, the mechanisms of mitochondrial toxicity include a decrease in mitochondrial membrane potential, inhibition of OXPHOS complex I and complex iv enzymes, decrease in oxygen consumption, and increased production of mitochondrial ROS [41]. A previous study showed that TDF caused a significant decrease of ATP in mice kidney and a decrease in succinate dehydrogenase activity, which is also an indication of the loss of inner mitochondrial membrane integrity. Moreover, TDF accumulation within proximal renal tubules led to mitochondrial injury and depletion [42][43]. Another study showed that long-term treatment with HAART causes mitochondrial dysfunction in HIV patients [44].
EFV, the most popular NNRTI, has been associated with metabolic disorders, hepatic toxicity and neurotoxicity [45][46]. EFV effects on the mitochondria include a decrease in mitochondrial membrane potential, inhibition of OXPHOS complex I enzymes, decrease in oxygen consumption, decrease in ATP production and increased production of mitochondrial ROS [47][48][49]. Dolutegravir (DTG) an important INSTI class drug alters mitochondrial function by decreasing ATP synthesis, depolarising the mitochondrial membrane, and has the potential to alter immunometabolism [33]. Another HAART toxicity mechanism is through an induction of oxidative stress [32].
Oxidative stress, a state of imbalance between oxidants production and antioxidants, and mitochondrial impairment result from xenobiotic metabolism and accompany one another [50]. Disruptions to mitochondrial function increase the production of ROS, mostly superoxide, through impaired OXPHOS [51]. Increased free radical production, over a period of time, depletes the antioxidant defense response, eventually resulting in oxidative damage to macromolecules including DNA, protein and lipid membranes [52]. NRTI, NNRTI and INTSI of HAART are linked with increased levels of oxidative stres and depletion of antioxidants in HIV-infected individuals (Figure 1).
HAART (TDF,FTC,DTG) treatment to primary rat microglia increased ROS levels [53]. TDF also significantly increased ROS production, depleted antioxidant GSH and the mitochondrial superoxide dismutase (MnSOD) [54]. HAART (3TC and DTG in combination with Abacavir) have been reported to induce liver toxicity through upregulation of ROS [55][56]. 3TC and FTC induced hepatotoxicity by triggering oxidative stress and depletion of the antioxidants GSH and SOD1 while also increasing the expression of ALT [57]. EFV-treated SweAPP N2a neurons displayed enhanced release of ROS [58]. Hamed, Aremu [59] and Ikekpeazu, Orji [44] showed that GSH and GPx levels were significantly reduced in rats subjected to HAART (TDF, 3TC and EFV) Ikekpeazu, Orji [44] further showed that HAART increased levels of MDA, which is the biomarker for oxidative stress and a by-product of lipid peroxidation. HAART-induced oxidative stress has been demonstrated to interfere with the mitochondrial function leading to reduction in GSH content [60][61]. Prolonged oxidative stress is reported to trigger inflammation, which is exacerbated by HAART usage.
3. HAART-Induced Chronic Inflammation and Insulin Resistance
HAART reduces systemic inflammation and immune activation, but not to levels synchronous with HIV-uninfected populations. Furthermore, over a prolonged period, HAART induces inflammation. With effective viral replication suppression by HAART, there is still a heightened pro-inflammatory condition in treated people compared to non-HAART consuming individuals. This develops to chronic and systemic inflammation, which, over time, promote pathophysiological metabolic complications [62][63][64].
A chronic inflammatory state is based on evidence of increased levels of various pro-inflammatory cytokines, including tumor necrosis factor alpha (TNF-α) [65][66][67], interleukin 1 beta (IL-1β), interleukin 6 (IL-6) [63][66], and biomarkers of inflammation such as nuclear factor kappa B (NF-κB) and C-reactive protein (CRP) [68]. The stimulation and release of pro-inflammatory mediators from one site promotes inflammation and usually ends up interfering and affecting other tissues, thereby amplifying the chronic inflammatory state, impairment of the cellular pathologies, and eventually tissue dysfunction/damage [69].
A recent study reported that HAART (TDF, FTC, and DTG) increased the mRNA levels of IL-1β, IL-6, and TNF-α in rats [53], as TDF modulated mitochondrial biogenesis and triggered inflammatory pathways. A recent study showed that TDF induced pro-inflammatory cytokines TNF-α and IL-1β in mice [70]. Ramamoorthy, Abraham [71] showed that the activation of NF-kB and its downstream pro-inflammatory target genes, inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and TNF-α, may play a critical role in the pathophysiology of TDF-induced renal damage in rats.
Hamed et al. (2021) reported that HAART (TDF, 3TC, and EFV) increased nitrite oxide (NO), a signaling molecule that plays a key role in the pathogenesis of inflammation. They also revealed that hepatic and renal membrane permeability, as well as caspase 3-dependent apoptosis, may be due to the stimulation of NF-kB and the enhancement of iNOS, essential factors of NO production. Moreover, it was reported that the oxidative stress induced by HAART may have triggered the inflammation (Figure 2). Oxidative stress can activate a variety of transcription factors, which lead to the differential expression of some of the genes involved in the inflammatory pathways [72][73]. Inflammation triggered by oxidative stress is the cause of many chronic diseases [74]. Chronic inflammation may cause pathophysiological complications such as insulin resistance [75][76][77].
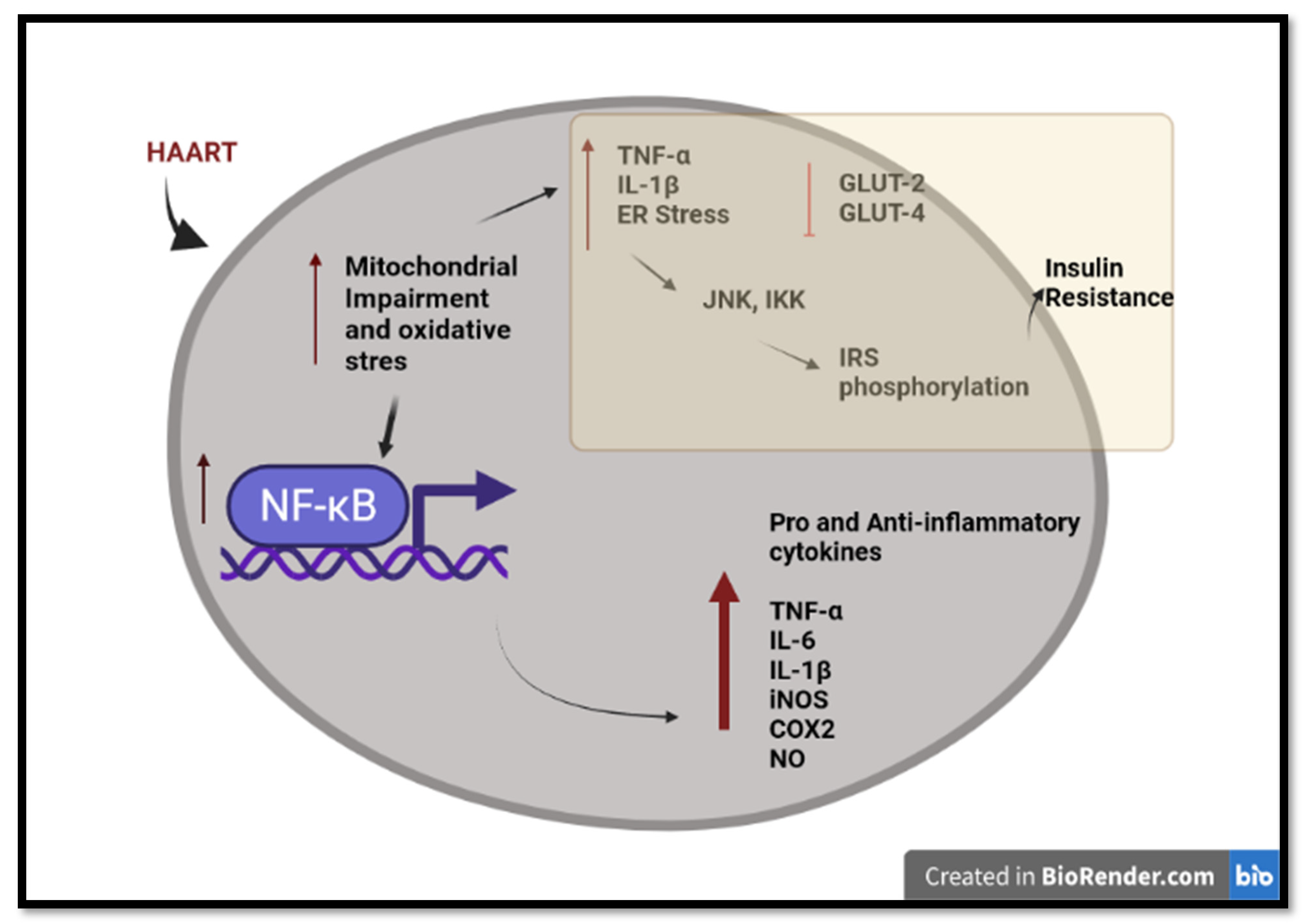
Figure 2. An overview of long-term HAART-induced inflammation and insulin resistance in HIV-infected individuals. HAART induced pro-inflammatory and anti-inflammatory cytokines through mitochondrial impairment, oxidative stress, and activation of NF-κB. HAART triggers insulin resistance via IRS phosphorylation and the inhibition of the glucose transporter. Created with BioRender.com (access date: 2 June 2022).
Increased TNF-α levels affect the insulin receptor substrate (IRS) proteins, leading to insulin resistance [78]. TNF-α induces activation of serine kinases such as cJun N-terminal kinase (JNK) and the two-kinase complex (IKKalpha and IKKbeta) (IKK), which phosphorylates IRS-1. The increased concentration of phosphorylated IRS-1 inhibits the insulin receptor, thus causing insulin resistance [79]. Lastly, there is compelling evidence that HAART inhibits insulin-stimulated glucose disposal via the blockade of glucose uptake by glucose transporter isoform 4 (GLUT 4) and glucose transporter isoform 2 (GLUT 2). This leads to insulin resistance and impaired β-cell function via down-regulation of the insulin receptors [80][81]. EFV has been shown to increase blood glucose levels to a greater degree, while DTG triggers the development of insulin resistance in human adipocytes [82].
The primary goal of HAART is to suppress HIV replication, thus allowing for immune reconstitution and subsequent longevity in HIV-infected individuals. The safety of these drugs is of paramount importance and should continuously be evaluated to achieve optimum adherence and the benefit of the therapy, while maintaining its efficacy. As a supplement, the application of adjuvants can benefit the HIV-infected population. Alternatively, the use of medicinal plants that may synchronously function with HAART and hence may minimize the toxic effects of HAART. Medicinal plants are one of the most important sources of novel nutritionally and pharmacologically active compounds, and have a well-documented history in the prevention and treatment of various diseases [83]. They contain many bioactive compounds that act to minimize oxidative stress and inflammation [84]. One such plant is Moringa oleifera (MO).
References
- WHO. Summary of the Global HIV Epidemic, 2020. World Heath Organisation. 2021. Available online: https://www.who.int/data/gho/data/themes/hiv-aids (accessed on 14 June 2022).
- Statssa. STATISTICAL RELEASE- P0302 Mid-Year Population Estimates, 2021. Department of statistics South Africa. 2021. Available online: http://www.statssa.gov.za/publications/P0302/P03022021.pdf (accessed on 10 June 2022).
- UNAIDS. Global AIDS Update 2021. UNAIDS Repots. 2021. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 14 June 2022).
- Volberding, P.A.; Deeks, S.G. Antiretroviral therapy and management of HIV infection. Lancet 2010, 376, 49–62.
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 1998, 338, 553–860.
- Watkins, C.C.; Treisman, G.J. Cognitive impairment in patients with AIDS—prevalence and severity. HIV/AIDS- Res. Palliat. Care 2015, 7, 35–47.
- Lee, G.Q.; Lichterfeld, M. Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr. Opin. HIV AIDS 2016, 11, 383–387.
- Durand, C.M.; Blankson, J.N.; Siliciano, R.F. Developing strategies for HIV-1 eradication. Trends Immunol. 2012, 33, 554–562.
- Politch, J.; Mayer, K.H.; Welles, S.L.; O’Brien, W.X.; Xu, C.; Bowman, F.; Anderson, D.J. Highly active antiretroviral therapy does not completely suppress HIV in semen of sexually active HIV-infected men who have sex with men. AIDS 2012, 26, 1535–1543.
- Falasca, K.; Reale, M.; Ucciferri, C.; Di Nicola, M.; Di Martino, G.; D’Angelo, C.; Coladonato, S.; Vecchiet, J. Cytokines, Hepatic Fibrosis, and Antiretroviral Therapy Role in Neurocognitive Disorders HIV Related. AIDS Res. Hum. Retrovir. 2017, 33, 246–253.
- Raposo, M.A.; Armiliato, G.N.D.A.; Guimarães, N.S.; Caram, C.A.; Silveira, R.D.D.S.; Tupinambás, U. Metabolic disorders and cardiovascular risk in people living with HIV/AIDS without the use of antiretroviral therapy. Rev. Soc. Bras. Med. Trop. 2017, 50, 598–606.
- Mohan, J.; Ghazi, T.; Chuturgoon, A.A. A Critical Review of the Biochemical Mechanisms and Epigenetic Modifications in HIV- and Antiretroviral-Induced Metabolic Syndrome. Int. J. Mol. Sci. 2021, 22, 12020.
- Ghislain, M.; Bastard, J.-P.; Meyer, L.; Capeau, J.; Fellahi, S.; Gérard, L.; May, T.; Simon, A.; Vigouroux, C.; Goujard, C.; et al. Late Antiretroviral Therapy (ART) Initiation Is Associated with Long-Term Persistence of Systemic Inflammation and Metabolic Abnormalities. PLoS ONE 2015, 10, e0144317.
- Hurwitz, B.E.; Klimas, N.G.; Llabre, M.M.; Maher, K.J.; Skyler, J.S.; Bilsker, M.S.; McPherson-Baker, S.; Lawrence, P.J.; LaPerriere, A.R.; Greeson, J.M.; et al. HIV, metabolic syndrome X, inflammation, oxidative stress, and coronary heart disease risk. Cardiovasc. Toxicol. 2004, 4, 303–315.
- Longenecker, C.T.; Jiang, Y.; Yun, C.-H.; Debanne, S.; Funderburg, N.T.; Lederman, M.M.; Storer, N.; Labbato, D.E.; Bezerra, H.G.; McComsey, G.A. Perivascular fat, inflammation, and cardiovascular risk in HIV-infected patients on antiretroviral therapy. Int. J. Cardiol. 2013, 168, 4039–4045.
- Ahmed, D.; Roy, D.; Cassol, E. Examining Relationships between Metabolism and Persistent Inflammation in HIV Patients on Antiretroviral Therapy. Mediat. Inflamm. 2018, 2018, 1–14.
- Nzuza, S.; Zondi, S.; Hurchund, R.; Owira, P.M. Highly Active Antiretroviral Therapy-Associated Metabolic Syndrome and Lipodystrophy: Pathophysiology and Current Therapeutic Interventions. J. Endocrinol. Metab. 2017, 7, 103–116.
- Hunt, P.W. HIV and Inflammation: Mechanisms and Consequences. Curr. HIV/AIDS Rep. 2012, 9, 139–147.
- Pérez, P.S.; Romaniuk, M.A.; Duette, G.A.; Zhao, Z.; Huang, Y.; Martin-Jaular, L.; Witwer, K.W.; Théry, C.; Ostrowski, M. Extracellular vesicles and chronic inflammation during HIV infection. J. Extracell. Vesicles 2019, 8, 1687275.
- White, A.J. Mitochondrial toxicity and HIV therapy. Sex. Transm. Infect. 2001, 77, 158–173.
- Feeney, E.R.; Mallon, P.W. Impact of mitochondrial toxicity of HIV-1 antiretroviral drugs on lipodystrophy and metabolic dysregulation. Curr. Pharm. Des. 2010, 16, 3339–3351.
- Nooka, S.; Ghorpade, A. HIV-1-associated inflammation and antiretroviral therapy regulate astrocyte endoplasmic reticulum stress responses. Cell Death Discov. 2017, 3, 17061.
- Ganta, K.K.; Chaubey, B. Mitochondrial dysfunctions in HIV infection and antiviral drug treatment. Expert Opin. Drug Metab. Toxicol. 2019, 15, 1043–1052.
- del MGutierrez, M.; GMateo, M.; Vidal, F.; Domingo, P. The toxicogenetics of antirretroviral therapy: The evil inside. J Curr. Med. Chem. 2011, 18, 209–219.
- Kolakowska, A.; Maresca, A.F.; Collins, I.J.; Cailhol, J. Update on Adverse Effects of HIV Integrase Inhibitors. Curr. Treat. Options Infect. Dis. 2019, 11, 372–387.
- Wang, Y.; De Clercq, E.; Li, G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin. Drug Metab. Toxicol. 2019, 15, 813–829.
- Post, F.A.; Sinxadi, P. Tenofovir disoproxil and renal mitochondrial toxicity: More studies in Africans are needed. AIDS 2022, 36, 1047–1048.
- Manda, K.R.; Banerjee, A.; Banks, W.A.; Ercal, N. Highly active antiretroviral therapy drug combination induces oxidative stress and mitochondrial dysfunction in immortalized human blood–brain barrier endothelial cells. Free Radic. Biol. Med. 2011, 50, 801–810.
- Chiao, S.K.; Romero, D.L.; Johnson, D.E. Current HIV therapeutics: Mechanistic and chemical determinants of toxicity. Curr. Opin. Drug Discov. Dev. 2009, 12, 53–60.
- Caron, M.; Auclair, M.; Vissian, A.; Vigouroux, C.; Capeau, J. Contribution of Mitochondrial Dysfunction and Oxidative Stress to Cellular Premature Senescence Induced by Antiretroviral Thymidine Analogues. Antivir. Ther. 2008, 13, 27–38.
- Nadezda, A.; Ana, B.-G.; Juan, V.E. Mitochondrial Toxicity in HAART: An Overview of In Vitro Evidence. Curr. Pharm. Des. 2011, 17, 2130–2144.
- Williams, A.A.; Sitole, L.J.; Meyer, D. HIV/HAART-associated oxidative stress is detectable by metabonomics. Mol. BioSyst. 2017, 13, 2202–2217.
- George, J.W.; Mattingly, J.E.; Roland, N.J.; Small, C.M.; Lamberty, B.G.; Fox, H.S.; Stauch, K.L. Physiologically Relevant Concentrations of Dolutegravir, Emtricitabine, and Efavirenz Induce Distinct Metabolic Alterations in HeLa Epithelial and BV2 Microglial Cells. Front. Immunol. 2021, 12, 639378.
- Maagaard, A.; Kvale, D. Long term adverse effects related to nucleoside reverse transcriptase inhibitors: Clinical impact of mitochondrial toxicity. Scand. J. Infect. Dis. 2009, 41, 808–817.
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J.R. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016, 39, 661–674.
- Birkus, G.; Hitchcock, M.; Cihlar, T. Assessment of mitochondrial toxicity in human cells treated with tenofovir: Comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2002, 46, 716–723.
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J Med Toxicol 2014, 10, 26–39.
- Dagan, T.; Sable, C.; Bray, J.; Gerschenson, M. Mitochondrial dysfunction and antiretroviral nucleoside analog toxicities: What is the evidence? Mitochondrion 2002, 1, 397–412.
- Gerschenson, M.; Nguyen, V.; Ewings, E.L.; Ceresa, A.; Shaw, J.A.; St. Claire, M.C.; Nagashima, K.; Harbaugh, S.W.; Harbaugh, J.W.; Olivero, O.A.; et al. Mitochondrial Toxicity in Fetal Erythrocebus patas Monkeys Exposed Transplacentally to Zidovudine Plus Lamivudine. AIDS Res. Hum. Retrovir. 2004, 20, 91–100.
- Samuels, R.; Bayerri, C.R.; Sayer, J.A.; Price, D.A.; Payne, B.A.I. Tenofovir disoproxil fumarate-associated renal tubular dysfunction: Noninvasive assessment of mitochondrial injury. AIDS 2017, 31, 1297–1301.
- Mccomsey, G.A.; Daar, E.S.; O’Riordan, M.; Collier, A.C.; Kosmiski, L.; Santana, J.L.; Fichtenbaum, C.J.; Fink, H.; Sax, P.E.; Libutti, D.E.; et al. Changes in Fat Mitochondrial DNA and Function in Subjects Randomized to Abacavir-Lamivudine or Tenofovir DF–Emtricitabine With Atazanavir-Ritonavir or Efavirenz: AIDS Clinical Trials Group Study A5224s, Substudy of A5202. J. Infect. Dis. 2012, 207, 604–611.
- Kohler, J.J.; Hosseini, S.H.; Hoying-Brandt, A.; Green, E.; Johnson, D.M.; Russ, R.; Tran, D.; Raper, C.M.; Santoianni, R.; Lewis, W. Tenofovir renal toxicity targets mitochondria of renal proximal tubules. Lab. Investig. 2009, 89, 513–519.
- Jafari, A.; Khalili, H.; Dashti-Khavidaki, S. Tenofovir-induced nephrotoxicity: Incidence, mechanism, risk factors, prognosis and proposed agents for prevention. Eur. J. Clin. Pharmacol. 2014, 70, 1029–1040.
- Ikekpeazu, J.E.; Orji, O.C.; Uchendu, I.K.; Ezeanyika, L.U. Mitochondrial and Oxidative Impacts of Short and Long-term Administration of HAART on HIV Patients. Curr. Clin. Pharmacol. 2020, 15, 110–124.
- Neukam, K.; Mira, J.A.; Ruiz-Morales, J.; Rivero, A.; Collado, A.; Torres-Cornejo, A.; Merino, D.; de Los Santos-Gil, I.; Macías, J.; González-Serrano, M.; et al. Liver toxicity associated with antiretroviral therapy including efavirenz or ritonavir-boosted protease inhibitors in a cohort of HIV/hepatitis C virus co-infected patients. J. Antimicrob. Chemother. 2011, 66, 2605–2614.
- Li, M.; Sopeyin, A.; Paintsil, E. Combination of Tenofovir and Emtricitabine with Efavirenz Does Not Moderate Inhibitory Effect of Efavirenz on Mitochondrial Function and Cholesterol Biosynthesis in Human T Lymphoblastoid Cell Line. Antimicrob. Agents Chemother. 2018, 62, e00691-18.
- Funes, H.A.; Blas-Garcia, A.; Esplugues, J.V.; Apostolova, N. Efavirenz alters mitochondrial respiratory function in cultured neuron and glial cell lines. J. Antimicrob. Chemother. 2015, 70, 2249–2254.
- Apostolova, N.; Blas-Garcia, A.; Galindo, M.J.; Esplugues, J.V. Efavirenz: What is known about the cellular mechanisms responsible for its adverse effects. Eur. J. Pharmacol. 2017, 812, 163–173.
- Schank, M.; Zhao, J.; Moorman, J.; Yao, Z. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174.
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077.
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110.
- Nagiah, S.; Phulukdaree, A.; Chuturgoon, A. Mitochondrial and Oxidative Stress Response in HepG2 Cells Following Acute and Prolonged Exposure to Antiretroviral Drugs. J. Cell. Biochem. 2015, 116, 1939–1946.
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Guo, M.-L.; Buch, S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells 2019, 8, 1168.
- Abraham, P.; Ramamoorthy, H.; Isaac, B. Depletion of the cellular antioxidant system contributes to tenofovir disoproxil fumarate—Induced mitochondrial damage and increased oxido-nitrosative stress in the kidney. J. Biomed. Sci. 2013, 20, 61.
- Wang, B.; Abbott, L.; Childs, K.; Taylor, C.; Agarwal, K.; Cormack, I.; Miquel, R.; Suddle, A. Dolutegravir-induced liver injury leading to sub-acute liver failure requiring transplantation: A case report and review of literature. Int. J. STD AIDS 2018, 29, 414–417.
- Christensen, E.S.; Jain, R.; Roxby, A.C. Abacavir/Dolutegravir/Lamivudine (Triumeq)–Induced Liver Toxicity in a Human Immunodeficiency Virus–Infected Patient. Open Forum Infect. Dis. 2017, 4, ofx122.
- Olaniyan, L.W.B.; Maduagwu, E.N.; Akintunde, O.W.; Oluwayelu, O.O.; Brai, B.I.C. Lamivudine-Induced Liver Injury. Open Access Maced. J. Med. Sci. 2015, 3, 545–550.
- Brown, L.; Jin, J.; Ferrell, D.; Sadic, E.; Obregon, D.; Smith, A.J.; Tan, J.; Giunta, B. Efavirenz Promotes β-Secretase Expression and Increased Aβ1-40,42 via Oxidative Stress and Reduced Microglial Phagocytosis: Implications for HIV Associated Neurocognitive Disorders (HAND). PLoS ONE 2014, 9, e95500.
- Hamed, M.; Aremu, A.; Akhigbe, R. Concomitant administration of HAART aggravates anti-Koch-induced oxidative hepatorenal damage via dysregulation of glutathione and elevation of uric acid production. Biomed. Pharmacother. 2021, 137, 111309.
- Sharma, B. Oxidative Stress in HIV Patients Receiving Antiretroviral Therapy. Curr. HIV Res. 2014, 12, 13–21.
- Mak, I.T.; Chmielinska, J.J.; Spurney, C.F.; Weglicki, W.B.; Kramer, J.H. Combination ART-Induced Oxidative/Nitrosative Stress, Neurogenic Inflammation and Cardiac Dysfunction in HIV-1 Transgenic (Tg) Rats: Protection by Mg. Int. J. Mol. Sci. 2018, 19, 2409.
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic Effects of Inflammation on Health during Chronic HIV Infection. Immunity 2013, 39, 633–645.
- Funderburg, N. Markers of coagulation and inflammation often remain elevated in ART-treated HIV-infected patients. Curr. Opin. HIV AIDS 2014, 9, 80–86.
- Gutierrez, M.d.M.; Mateo, M.G.; Vidal, F.; Domingo, P. Does choice of antiretroviral drugs matter for inflammation? Expert Rev. Clin. Pharmacol. 2019, 12, 389–396.
- Kalayjian, R.C.; Machekano, R.N.; Rizk, N.; Robbins, G.K.; Gandhi, R.T.; Rodriguez, B.A.; Pollard, R.B.; Lederman, M.M.; Landay, A. Pretreatment Levels of Soluble Cellular Receptors and Interleukin-6 Are Associated with HIV Disease Progression in Subjects Treated with Highly Active Antiretroviral Therapy. J. Infect. Dis. 2010, 201, 1796–1805.
- Hunt, P.W.; Sinclair, E.; Rodriguez, B.; Shive, C.; Clagett, B.; Funderburg, N.; Robinson, J.; Huang, Y.; Epling, L.; Martin, J.N.; et al. Gut Epithelial Barrier Dysfunction and Innate Immune Activation Predict Mortality in Treated HIV Infection. J. Infect. Dis. 2014, 210, 1228–1238.
- Tenorio, A.R.; Chan, E.S.; Bosch, R.J.; Macatangay, B.J.C.; Read, S.W.; Yesmin, S.; Taiwo, B.; Margolis, D.M.; Jacobson, J.M.; Landay, A.L.; et al. Rifaximin has a marginal impact on microbial translocation, T-cell activation and inflammation in HIV-positive immune non-responders to antiretroviral therapy—ACTG A5286. J. Infect. Dis. 2015, 211, 780–790.
- Lumeng, C.N. Innate immune activation in obesity. Mol. Asp. Med. 2013, 34, 12–29.
- Hirabara, S.M.; Gorjão, R.; Vinolo, M.A.; Rodrigues, A.C.; Nachbar, R.T.; Curi, R. Molecular Targets Related to Inflammation and Insulin Resistance and Potential Interventions. J. Biomed. Biotechnol. 2012, 2012, 379024.
- Fields, J.A.; Swinton, M.K.; Carson, A.; Soontornniyomkij, B.; Lindsay, C.; Han, M.M.; Frizzi, K.; Sambhwani, S.; Murphy, A.; Achim, C.L.; et al. Tenofovir disoproxil fumarate induces peripheral neuropathy and alters inflammation and mitochondrial biogenesis in the brains of mice. Sci. Rep. 2019, 9, 17158.
- Ramamoorthy, H.; Abraham, P.; Isaac, B.; Selvakumar, D. Role for NF-κB inflammatory signalling pathway in tenofovir disoproxil fumarate (TDF) induced renal damage in rats. Food Chem. Toxicol. 2017, 99, 103–118.
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616.
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114.
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797.
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564.
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409.
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480.
- Méndez-García, L.A.; Trejo-Millán, F.; Martínez-Reyes, C.P.; Manjarrez-Reyna, A.N.; Esquivel-Velázquez, M.; Melendez-Mier, G.; Islas-Andrade, S.; Rojas-Bernabé, A.; Kzhyshkowska, J.; Escobedo, G. Infliximab ameliorates tumor necrosis factor-alpha-induced insulin resistance by attenuating PTP1B activation in 3T3L1 adipocytes in vitro. Scand. J. Immunol. 2018, 88, e12716.
- Pedro, M.N.; Rocha, G.Z.; Guadagnini, D.; Santos, A.; Magro, D.O.; Assalin, H.B.; Oliveira, A.G.; Pedro, R.D.J.; Saad, M.J.A. Insulin Resistance in HIV-Patients: Causes and Consequences. Front. Endocrinol. 2018, 9, 514.
- Hruz, P.W. Molecular Mechanisms for Altered Glucose Homeostasis in HIV Infection. Am. J. Infect. Dis. 2006, 2, 187–192.
- Avari, P.; Devendra, S. Human immunodeficiency virus and type 2 diabetes. Lond. J. Prim. Care 2017, 9, 38–42.
- Gorwood, J.; Bourgeois, C.; Pourcher, V.; Pourcher, G.; Charlotte, F.; Mantecon, M.; Rose, C.; Morichon, R.; Atlan, M.; Le Grand, R.; et al. The Integrase Inhibitors Dolutegravir and Raltegravir Exert Proadipogenic and Profibrotic Effects and Induce Insulin Resistance in Human/Simian Adipose Tissue and Human Adipocytes. Clin. Infect. Dis. 2020, 71, e549–e560.
- Van Wyk, B.-E.; Wink, M. Medicinal Plants of the World; CABI: Wallingford, UK, 2018.
- Zhang, L.; Ravipati, A.S.; Koyyalamudi, S.R.; Jeong, S.C.; Reddy, N.; Smith, P.T.; Bartlett, J.; Shanmugam, K.; Münch, G.; Wu, M.J. Antioxidant and Anti-inflammatory Activities of Selected Medicinal Plants Containing Phenolic and Flavonoid Compounds. J. Agric. Food Chem. 2011, 59, 12361–12367.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
08 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No