 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Frederique Souaze | + 3901 word(s) | 3901 | 2020-10-20 11:19:44 | | | |
| 2 | Bruce Ren | Meta information modification | 3901 | 2020-10-27 04:47:47 | | |
Video Upload Options
In this review, we focus on how CAFs participate in cancer progression by modulating cancer cells metabolic functions and mitochondrial apoptosis.Resistance of solid cancer cells to chemotherapies and targeted therapies is not only due to the mutational status of cancer cells but also to the concurring of stromal cells of the tumor ecosystem, such as immune cells, vasculature and cancer-associated fibroblasts (CAFs). The reciprocal education of cancer cells and CAFs favors tumor growth, survival and invasion. Mitochondrial function control, including the regulation of mitochondrial metabolism, oxidative stress and apoptotic stress are crucial for these different tumor progression steps. In this review, we focus on how CAFs participate in cancer progression by modulating cancer cells metabolic functions and mitochondrial apoptosis.
1. Introduction
Mitochondria have been implicated in tumoral progression since Otto Warburg described mitochondrial dysfunction associated with glycolytic activity increase even under normoxia as a tumor promoter in 1927 [1]. Since then, it has been shown that mitochondria, even impaired, still provide malignant cells with energy and biosynthetic precursors, and control redox homeostasis and resistance to apoptosis. Indeed, the intrinsic pathway of apoptosis relies on mitochondrial outer membrane permeabilization (MOMP) leading to caspases activation and subsequent loss of cell integrity. Thus, the mitochondrial apoptosis resistance process taking place up or downstream of MOMP is crucial to cancer cell survival.
Cancer cell interactions with others cell types, such as cancer-associated fibroblasts (CAFs), immune cells and endothelial cells, actively participate in tumor progression, including tumor growth, survival and invasion [2]. In particular, CAFs and tumor cells strongly dialogue via soluble factors, exosomes, extracellular matrix components and direct contacts [3]. The two cell types educate each other to adapt to their nutritional and signaling environment. Glycolytic CAFs have been shown to enhance the contribution of mitochondria to energy production and biogenesis in cancer cells, also promoting tumor progression. This process was called the “Reverse Warburg Effect” [4]. Here, we focus on both mitochondrial metabolic activity and the apoptosis resistance of cancer cells under CAFs control. Importantly, the metabolic dialogue between CAFs and cancer cells implies a reciprocal influence of cancer cells on CAFs metabolism, which participates in their pro-tumoral effects. Moreover, cancer cells have been shown to attract and activate fibroblasts via cytokines and growth factors [5].
2. CAFs Sustain Cancer Cells Mitochondria
2.1. CAFs Reorganize Cancer Cells’ Mitochondrial Metabolism
Here, we focus on CAF/cancer cell metabolic interactions that impact malignant cells’ mitochondria.
CAFs have been shown to fuel cancer cells with organic and amino acids. Pyruvate is an organic acid at the crossroad between glycolysis and mitochondrial oxidative phosphorylation (OXPHOS). It fuels the tricarboxylic acid (TCA) cycle and subsequent mitochondrial respiration. CAFs can directly provide cancer cells with pyruvate (as shown in lymphoma [6]), and also indirectly by providing lactate (as shown in prostate cancer [7][8] and breast cancer [9]) or alanine (as shown in pancreatic cancer [10]), both latter metabolites being transformed into pyruvate via active lactate dehydrogenase and alanine aminotransferase, respectively. CAFs also fuel malignant cells with glutamine in glutamine-deprived conditions (as shown in ovarian cancer [11]), which is transformed into glutamate and then alpha-ketoglutarate to enter the TCA cycle and generate biosynthetic precursors. Of note, metabolites are not only exchanged from CAFs to cancer cells via their soluble forms since amino-acids and TCA cycle intermediates can be shuttled via exosomes, upregulating, in this case, glycolysis but reducing OXPHOS (as in prostate and pancreatic cancer cells [12]).
Thus, CAFs provide intermediate metabolites for malignant cells mitochondrial activity. More precisely, these metabolites fuel malignant cells’ TCA cycle, which feeds biosynthetic pathways to produce key precursors such as lipids, proteins and nucleic acids, thus promoting primary and metastatic cell growth [10][11]. In some of the studies, TCA cycle modulation induced by CAFs even leads to higher malignant cell oxygen consumption, reflecting mitochondrial respiration increase [10]. In addition, a CAFs-induced increase in TCA cycle activity is associated with primary patient malignant cell survival. Of note, CAF-induced metabolite consumption is enabled by the concomitant upregulation of metabolic transporters, such as lactate transporter MCT1 (in prostate cancer cells [13]).
Beside fueling TCA, lactate promotes mitochondrial biogenesis. Indeed, lactate consumption by metastatic prostate cancer cells under CAFs-control, via shifting NAD+/NADH cell equilibrium toward NAD+ that is a substrate of Sirtuins (SIRTs) [14], activates SIRT1/PGC1α axis that promotes mitochondrial biogenesis and activity. CAFs might also favor cancer stem cell traits as SIRT1 has been shown to regulate cell stemness [15], and as this phenotype mainly relies on oxidative phosphorylation (in ovarian and breast cancer [16][17]). Moreover, SIRT1/PGC1α axis is amplified by concomitant activation of proto-oncogene tyrosine-protein kinase Src due to TCA cycle deregulation-induced reactive oxygen species (ROS) production. Interestingly, ROS production, which is elicited by both respiratory chain overload and mild respiration dysfunction, has been shown to induce Src activation, promoting tumor cell migration [18]. Mitochondrial ROS, related to CAF-induced metabolic reprogramming, could be involved in many other tumor progression mechanisms, since sustained ROS production promotes tumor proliferation, genetic instability and some treatments resistance [19].
Upregulated mitochondrial activity, associated with the downregulation of TCA cycle enzymes, can also lead to the accumulation of mitochondrial metabolites, called oncometabolites, when participating in tumor progression. For instance, CAF-induced mitochondrial fueling of prostate malignant cells leads to succinate and fumarate accumulation [8]. Importantly, succinate accumulation induces HIF1α stabilization and subsequent oncogenic epithelial–mesenchymal transition (EMT). Fumarate accumulation has also been shown to favor EMT of renal cancer cells via epigenetic modifications [20], thus promoting invasion.
Some CAFs subpopulations have also been shown to reorganize cancer cell metabolism via secreting cytokines that favor glycolysis and TCA cycle intermediates production, resulting in tumor growth and invasion in breast and pancreatic cancers [21]. A similar cytokine-based interaction was shown in ovarian cancer, implying IL-6, CXCL-5 and CXCL-10, which promotes self-stored glycogen utilization by cancer cells to fuel glycolysis and subsequent mitochondrial activity [22]. CAFs’ metabolic impact on cancer cells is summarized in Figure 1.
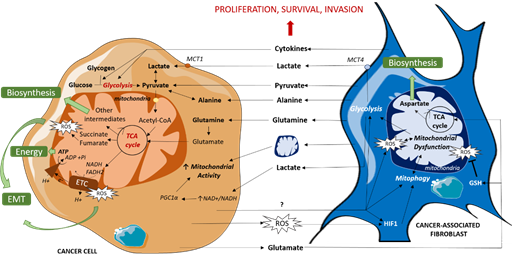
Figure 1. Metabolic dialogue between cancer cells and CAFs. CAFs fuel TCA cycle by directly providing cancer cells with organic (lactate, pyruvate) and amino acids (alanine, glutamine) or indirectly by enhancing glycolysis via cytokine release, resulting in an increase in mitochondrial activity which leads to energy and biosynthetic precursor production and redox state modulation. CAFs also enhance mitochondrial activity via mitochondrial transfers. In turn, cancer cells induce mitochondrial dysfunction in CAFs, mitophagy and ROS production, amplifying their mutual support. EMT: Epithelial Mesenchymal Transition, ETC: Electron Transport Chain, GSH: reduced glutathione, mtDNA: Mitochondrial DNA, MCT: MonoCarboxylate Transporter, ROS: Reactive Oxygen Species, TCA cycle: TriCarboxylic Acid cycle.
2.2. CAFs Provide Intact Mitochondria to Support Cancer Cells Mitochondrial Activity
Mitochondrial transfers occur between cancer cells and different cell types and are thought to happen in many cancers to optimize or repair the malignant cells’ metabolic machinery [23], promoting cancer progression. Indeed, mtDNA transfer from in vivo local environment cells to mitochondrial deficient metastatic mammary and melanoma tumor cells in syngenic murine models was shown to restore the respiration of primary and metastatic tumor cells [24]. High relative mtDNA copy number resulting in a high bioenergetic mitochondrial function has been shown to confer an advantage for tumor invasion [25]. Active transfers of mitochondria from human mesenchymal stem cells (MSCs) and skin fibroblasts have also been shown to restore the mitochondrial network of mitochondrial deficient lung adenocarcinoma epithelial cells [26], and mitochondria uptake from MSCs has been shown to promote breast cancer cells OXPHOS and proliferation as well as invasion [27]. Moreover, mitochondrial transfers from bone-marrow MSCs through the endocytic pathway protect leukemia initiating cells potential from ROS-inducing chemotherapy in acute myeloid leukemia [28]. Similar transfers from endothelial cells protect breast and ovarian tumor cells from doxorubicin-induced cell death in vitro [29].
CAFs also reshape mitochondrial network and genome expression in cancer cells. Indeed, primary CAFs have been shown to transfer mitochondria to cancer cells via cellular bridges, also called tunneling nanotubes, in prostate cancer and acute lymphoblastic leukemia (ALL) [30]. Horizontal transfer of functional mitochondria from CAFs enhances prostate cancer cells mitochondrial mass and activity, thus fostering lactate-fueled respiration and further promoting malignancy. In this study, malignant cells that are pre-incubated with CAFs conditioned media are more prone to receive mitochondria, suggesting that a prior education by CAFs is needed in this kind of interaction. Moreover, the transfer of mitochondrial mass in ALL cells from CAFs generated from primary MSCs under ROS-inducing chemotherapy protects cancer cells against the same ROS-inducing agent and is reversed by microtubule inhibition in vivo [30].
Although their frequency and triggering signals need to be further studied, such mitochondrial transfers from CAFs to cancer cells (Figure 1) support the idea of a strong and multifaceted interaction between the two cell types based on mitochondrial processing that promotes tumor malignancy.
2.3. CAFs Protect Cancer Cells Mitochondrial Integrity by Regulating Pro- and Anti-Apoptotic Proteins
CAFs also protect cancer cells from chemotherapies and specific pro-apoptotic drugs via the modulation of mitochondrial apoptosis-related proteins, favoring tumor escape and proliferation (Figure 2).
Stress stimuli, such as DNA damage or oxidative stress, can trigger BAX and/or BAK oligomerization at the mitochondrial membrane, resulting in mitochondrial outer membrane permeabilization (MOMP). This leads to the cytosolic release of cytochrome c, which induces caspases activation and consequent apoptosis. Stress stimuli alter the equilibrium between pro- and anti-apoptotic BCL-2 family proteins involved in the regulation of BAX and BAK oligomerization. The anti-apoptotic proteins BCL-2, MCL-1 and BCL-xL prevent MOMP via direct interactions with the pro-apoptotic proteins and have been shown to protect cancer cells from stress stimuli induced by chemotherapies [31]. CAFs favor chemoresistance by modulating some BCL-2 family anti-apoptotic protein levels in malignant cells. Indeed, under cisplatin treatment, CAFs promote the phosphorylation and activation of STAT3, which upregulates the levels of BCL-2 in ovarian cancer [32] and in lung adenocarcinoma triggered by IL11 paracrine signaling [33], thus resulting in chemoresistance. CAFs have also been shown to enhance BCL-2 protein levels in bladder cancer cells via the activation of IGF1/ERβ signaling in cancer cells in vitro and in vivo [34]. Moreover, our team showed that CAFs protect luminal breast cancer cells from apoptosis by upregulating the anti-apoptotic MCL-1 in cancer cells via a paracrine IL-6 signaling, which triggers ERK phosphorylation [35]. CAFs-induced MCL-1 upregulation has also been shown to protect breast cancer cells from apoptosis during cell detachment, also known as anoïkis. Indeed, CAFs-secreted IGF-binding proteins trigger the ERK/MAPK pathway in cancer cells and the subsequent inhibition of GSK3 that normally induces MCL-1 degradation [36]. To a larger extent, CAFs have been shown to render HER2+ breast cancer cells less sensitive to apoptosis [37]. In this study, the regulation of apoptotic threshold is implicated in CAFs protective effects to lapatinib, an EGFR/HER2 inhibitor. Notably, the elevation of apoptotic threshold implicates the JAK/STAT signaling pathway in both carcinoma cells and CAFs.
CAFs are a major source of extracellular matrix (ECM) components, such as type I collagen, fibronectin and laminin. MCL1 level has been shown to be upregulated in pancreatic cancer cells cultured on the type I collagen matrix, compared to plastic, conferring resistance to antimetabolite 5-fluorouracil [38]. Moreover, integrins expressed at cancer cells surface trigger survival signals when ligating to ECM components. Indeed, the decrease in BCL-2 protein level induced by paclitaxel treatment is blocked by the integrin-mediated cell attachment of cancer cells to collagen I, fibronectin or laminin, although the induction of signals depends on the cell lines [39]. These results suggest that CAFs could also modulate BCL-2 family proteins in cancer cells via ECM secretion. Interestingly, CAFs conditioned media have been shown to induce the expression of some integrins and BCL-2 in lung carcinoma cells [40]. Notably, integrin B1 and B3 overexpression in malignant cells promotes BCL-2 expression in this study. The mechanical force induced by CAFs-secreted ECM could also be implicated in anti-apoptotic proteins regulation since yes-associated protein (YAP), which can be regulated by mechanical forces [41], has been shown to promote BCL-2 expression in oral squamous carcinoma cells [42].
CAFs also protect cancer cells from chemotherapies by acting downstream of MOMP. Müerköster and colleagues have shown that etoposide resistance of pancreatic cancer cells induced by CAFs in a co-culture model does not rely on pro- nor anti-apoptotic BCL2 family protein regulation [43]. Instead, CAFs epigenetically downregulate caspase expression, inducing transcription factor STAT1, thus limiting caspases 9, 3 and 7 activation. In in vitro and in vivo lung cancer models, CAFs secretion of Annexin A3 has been shown to stimulate cancer cells survivin, known to inhibit caspases activity, thus leading to the decrease in caspases 3 and 8 cleavage under cisplatin treatment [44]. Similarly, CAFs protect lung and ovarian cancer cells from cisplatin via increasing survivin by promoting STAT3 phosphorylation. CAFs also protect pancreatic cancer cells from gemcitabine by inducing survivin expression [45]. ECM also acts downstream of MOMP since laminin upregulates survivin by inducing focal adhesion kinase phosphorylation in pancreatic cancer cells, thus promoting chemoresistance [46].
Thus, CAFs have been shown to modulate the expression and activity of anti-apoptotic proteins of the BCL-2 family in cancer cells, resulting in drug resistance. Interestingly, these proteins have been shown to modulate mitochondrial metabolic function in different cell models. Indeed, BCL-2 promotes mitochondrial respiration in cancer cells, resulting in a pro-oxidant state in basal conditions [47], while BCL-xL stabilizes the inner membrane potential and thus modulates mitochondrial energetics in neurons [48]. Moreover, such non-canonical functions of the BCL-2 family anti-apoptotic proteins can depend on their location, where they can interact with metabolic enzymes and transporters. For instance, MCL-1 interaction with outer mitochondrial membrane voltage-dependent anion channel (VDAC) has been shown to increase mitochondrial Ca2+ uptake and reactive oxygen species generation in lung cancer cells [49]. MCL-1 located at the mitochondrial matrix has also been shown to induce respiration in a mouse embryonic fibroblastic model [50]. BCL-xL can also interact with VDAC to favor the open configuration of the channel and metabolite passage through the outer mitochondrial membrane in a murine pro-B lymphocytic cell line [51]. Furthermore, BCL-xL increases adenosine triphosphate ATP production within mitochondria by interacting with the β subunit of F(1)F(0) ATP synthase in neurons [52]. These studies show non-canonical functions of anti-apoptotic BCL-2, BCL-xL and MCL-1, related to mitochondrial function. One can suggest an interconnection between the regulation of mitochondrial function and the regulation of expression or activity of anti-apoptotic proteins of cancer cells in the pro-tumoral effects of CAFs. In particular, it would be interesting to establish whether CAFs modulate BCL-2 anti-apoptotic protein location and interactome with metabolic enzymes and transporters.
3. Mitochondrial Processing in CAFs Is Implicated in Their Pro-Tumoral Effects in an Ecosystemic Context
As seen above, CAFs participate in tumor progression via their ability to modulate mitochondrial activity. In this section, we report that this ability relies in part on mitochondrial stress of CAFs triggered by cancer cells, highlighting the reciprocal education between the two cell types. Importantly, mitochondrial processing in fibroblasts participates in their activation. We also discuss the heterogeneity of mitochondrial-mediated interactions between CAFs and cancer cells.
3.1. CAFs Mitochondrial Activity Is under Cancer Cells Control
Reciprocal education between CAFs and cancer cells is given in part by their metabolic crosstalk. Glycolytic switch in CAFs which mediates cancer cells metabolic changes is regulated by cancer cells themselves (Figure 1). Indeed, prostate cancer cells induce glycolytic switch in CAFs via the downregulation of mitochondrial deacetylase SIRT3 that promotes oxidative stress and HIF1 stabilization. Oral squamous cell carcinoma cells have also been shown to metabolically reprogram normal oral fibroblasts in an indirect co-culture model by inducing mitochondrial dysfunction reported as ROS accumulation, mitochondrial permeability transition pore opening, hypoxia and mitophagy, associated with an increase in aerobic glycolysis [53]. Moreover, breast cancer cells have been shown to favor CAF oxidative stress via hydrogen peroxide secretion, leading to CAF autophagy and mitophagy mediated by HIF1 stabilization, and promoting mitochondrial dysfunction and enhanced glycolysis [54]. A recent study has shown that triple negative breast cancer cells can induce CAF glycolytic switch and mitophagy via exosome-mediated integrin ITGB4 export that induces ITGB4 expression by CAFs themselves [55]. In these studies, cancer-cell-triggered glycolytic CAFs secrete lactate. Importantly, this secretion is promoted by the upregulation of the monocarboxylate transporter MCT4 in CAFs [53][55].
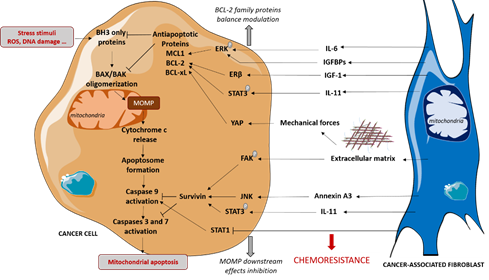
Figure 2. CAFs-mediated protection of cancer cells to mitochondrial apoptosis. CAFs induce chemoresistance in cancer cells by protecting them from mitochondrial apoptosis both by regulating anti-apoptotic proteins level and by limiting caspases activation. These effects are mediated by secretion of cytokines and growth factors and by extracellular matrix production, that is a feature of CAFs. IGF-1: Insulin-like Growth Factor-1, IGFBP: Insulin-like Growth Factor Binding Protein, IL: Interleukin, MOMP: mitochondrial Outer Membrane Permeabilization.
Cancer cells, via mitochondrial processing, modulate other CAFs metabolic features involved in their pro-tumoral effects. Under stiff matrix conditions, squamous cell carcinoma cells have been shown to secrete glutamate that can be used by CAFs to fuel the TCA cycle and produce aspartate, that, in turn, fuels cancer cells for nucleotides biosynthesis, favoring tumor growth [56]. Notably, cancer-cell-secreted glutamate induces the glutathione pathway in CAFs, thus limiting the accumulation of ROS and superoxide induced by matrix stiffness. In a glutamine deprived co-culture ovarian cancer model, malignant cells have been shown to enhance TCA cycle activity in CAFs to maintain glutamate and citrate levels for glutamine synthesis, that is then secreted by CAFs to support cancer cells proliferation.
Altogether, CAFs metabolism that is involved in cancer cells mitochondrial processing is controlled by cancer cells themselves within a reciprocal mitochondrial education.
3.2. Mitochondrial Dynamics Is Involved in Fibroblasts Activation
Although CAFs can arise from bone marrow-derived precursors, mesenchymal stem cells, or endothelial cells, resident fibroblasts have been described as the major source of CAFs [57].
Mitochondrial activity and dynamics have been particularly implicated in TGF-β signaling leading to fibroblast activation into myofibroblasts, characterized by αSMA expression and invasive and migratory abilities. In normal human lung fibroblasts, mitochondrial ROS generated at mitochondrial respiratory chain complex III are required for TGF-β-induced gene expression, in particular, αSMA [58]. High mitochondrial generation of ROS caused by respiratory chain complex I dysfunction also correlates with myofibroblast activation [59]. TGF-β is not the only activation signaling modulated by mitochondria, since mitochondrial ROS have also been shown to regulate PDGF-induced signaling in primary mouse embryonic fibroblasts via oxidation of protein tyrosine phosphatases [60], possibly promoting fibroblast activation. Moreover, mitochondrial dynamics are involved in TGF-β signaling, since targeting mitochondrial-fission-mediator DRP-1 inhibits TGF-β-induced rat kidney fibroblast cell activation [61].
Interestingly, the acquisition of CAFs’ metabolic characteristics by normal fibroblasts co-cultured with malignant cells precedes the acquisition of the fibroblast activation protein (FAP) and loss of Caveolin 1. This result suggests that metabolic reprogramming could participate in the activation of fibroblasts by cancer. Concordant with this, TGF-β-induced early increase in glycolysis in lung fibroblasts sustains transformation into myofibroblasts. More precisely, glycolysis increases the TCA cycle intermediate succinate, which stabilizes HIF1α and promotes myofibroblastic differentiation [62], thus implicating metabolic mitochondrial function in TGF-β-induced fibroblast differentiation. Reduced mitochondrial α-ketoglutarate has also been shown to stabilize HIF1α under normoxia in human colon CAFs under TGF-β or PDGF stimulation, thus favoring glycolysis. Of note, albeit no consensus was found, HIF1α has been shown in several studies to permit oncogenic gain of functions in fibroblasts [63]. These results highlight the essential role of mitochondria in fibroblast activation.
Thus, fibroblast mitochondrial activity participates in the signaling pathways, leading to their activation. Within tumors, the fibroblast metabolic switch could either be an active phenomenon favoring their activation or a consequence of their activated state.
3.3. Mitochondrial-Mediated Interactions between CAFs and Cancer Cells Are Heterogeneous
CAFs exert specific metabolic and mitochondrial processing in cancer cells depending on cancer types and subtypes. As described earlier, the nature of the identified secreted nutrients and subsequent metabolism seems to vary according to the localization of the cancer—lactate in prostate cancer, lactate/pyruvate in breast cancer, alanine in pancreatic cancer and glutamine in ovarian cancer. This heterogeneity could originate from the specific metabolism of cancer cells [64], that could be a factor of strong interaction between cancer cells and CAFs. Additional to these disparities between cancer, depending on the tissue they originate from, intra-tumoral heterogeneity could be a source of varying cancer cells/CAFs interactions. As previously reviewed by Strickaert and colleagues [65], the concept of tumor heterogeneity includes the diversity of the cell populations, including stromal cells, the cell location within the tumor, the epigenetic and genetic effects in cancer cells over time and, eventually, the variation in metabolism, all of these being strikingly linked to each other. Concerning cell location within the tumor, by using a micro-patterned co-culture model consisting in a breast cancer cell (MCF7) island surrounded by stromal cells, it has been shown that stromal mechanical constraints induce spatial heterogeneity of mitochondrial activities in cancer cells, with an impact on metabolism and the metastatic potential of cancer cells [66]. On the other hand, in melanoma, it has been reported that heterogeneity in MCT1-high and MCT1-negative or low expressing cancer cells discriminate their metastatic potential. MCT1-high cells uptake more lactate and are more efficient to metastasize, implying that glycolytic CAFs exert a differential pro-metastatic effect on these melanoma cell sub-populations [67]. Moreover, metabolic heterogeneity has been shown within malignant cells of mammary tumors by single-cell transcriptomics in a MMTV-PyMT mouse model [68]. Indeed, one PyMT cell subpopulation expresses higher levels of genes involved in OXPHOS, while another shows higher glycolytic process gene expression. Intra-tumoral malignant cell metabolic heterogeneity could either be the result of the interactions with CAFs or could directly modulate this interaction, resulting in different pro-tumoral effects. In human breast cancer, metabolic interaction between cancer and stromal cells could further vary according to molecular subtype [69]. Indeed, this immunohistochemical study suggests a correlation between the subtype and the metabolic phenotype of the tumor (Warburg type with glycolytic tumor cells and non-glycolytic stroma or Reverse Warburg type with non-glycolytic tumor cells and glycolytic stroma). Moreover, the secreted intermediates that modulate mitochondrial function are specific of certain subtypes of cancer. It seems the case with breast-cancer-cell-secreted ITGB4 that it is mainly secreted by triple negative breast cancer cell lines, and more specifically by some of the lines of this molecular subtype.
The heterogeneity of the mitochondrial-mediated dialogue between CAFs and cancer cells can also be highlighted by the heterogeneity of mitochondrial activity of CAFs within tumor. CAFs are indeed heterogeneous within tumors, which could be explained by their adaptability to their environment. Determining whether CAFs mitochondrial activity also depends on their diverse cellular origins [70] would be of particular interest. Costa and colleagues identified four subsets of CAFs within breast tumors from patients [71]. RNA sequencing shows that one subset, called S4 and characterized among others by high αSMA expression and low FAP expression, exhibits gene enrichment in oxidative metabolism. The four subsets have also been identified in ovarian cancer [72]. In this latter model, the S4 subset exhibits a strong enrichment in genes encoding electron transport chain proteins. Moreover, Qian and colleagues recently identified metabolic heterogeneity between CAFs subpopulations commonly found in colorectal, ovarian and lung tumors, with some populations characterized by glycolytic signature, based on single-cell analysis of transcription factor activities [73]. These studies suggest heterogeneity in CAFs mitochondrial function within the tumor. Costa and colleagues also show specific spatial distribution with subsets S1 and S4 preferentially accumulating in the tumor while the other subtypes are found in juxta-tumors. Of note, CAFs present in juxta-tumors are enriched in CAFs with genes involved in oxidative stress, potentially revealing mitochondrial dysfunction. These results suggest that spatial proximity to the tumor could be important for CAF mitochondrial function. CAFs have indeed been shown to adapt their metabolism to the nutritional context [74], that is mainly influenced by cancer cells metabolic activity.
Thus, the interactions between CAFs and cancer cells and the consequences on mitochondrial functions depend on cancer type and seem heterogeneous within tumors, emphasizing the complexity of the understanding of the dialogue between the two cell types.
References
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530, doi:10.1085/jgp.8.6.519.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437, doi:10.1038/nm.3394.
- Elenbaas, B.; Weinberg, R.A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 2001, 264, 169–184, doi:10.1006/excr.2000.5133.
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001, doi:10.4161/cc.8.23.10238.
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619, doi:10.1007/s00441-016-2467-x.
- Sakamoto, A.; Kunou, S.; Shimada, K.; Tsunoda, M.; Aoki, T.; Iriyama, C.; Tomita, A.; Nakamura, S.; Hayakawa, F.; Kiyoi, H. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 2019, 110, 269–278, doi:10.1111/cas.13873.
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; de Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res. 2012, 72, 5130–5140, doi:10.1158/0008-5472.CAN-12-1949.
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355, doi:10.1038/s41388-019-0805-7.
- Sun, K.; Tang, S.; Hou, Y.; Xi, L.; Chen, Y.; Yin, J.; Peng, M.; Zhao, M.; Cui, X.; Liu, M. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine 2019, 41, 370–383, doi:10.1016/j.ebiom.2019.02.025.
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483, doi:10.1038/nature19084.
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700, doi:10.1016/j.cmet.2016.10.011.
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250, doi:10.7554/eLife.10250.
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783, doi:10.4161/cc.10.11.15659.
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295, doi:10.1146/annurev.pathol.4.110807.092250.
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218, doi:10.1111/acel.12685.
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319, doi:10.18632/oncotarget.2010.
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyari, Y.; Dratver, M.B.; Pajonk, F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014, 146, 525–534, doi:10.1007/s10549-014-3051-2.
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; de Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766, doi:10.1016/j.celrep.2014.06.043.
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110, doi:10.1016/j.drup.2004.01.004.
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547, doi:10.1038/nature19353.
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; Delgado, B.D.L.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290, doi:10.1038/s41467-020-15104-3.
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155, doi:10.1016/j.cmet.2018.08.007.
- Rebbeck, C.A.; Leroi, A.M.; Burt, A. Mitochondrial Capture by a Transmissible Cancer. Science 2011, 331, 303–303, doi:10.1126/science.1197696.
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94, doi:10.1016/j.cmet.2014.12.003.
- Lin, C.S.; Lee, H.T.; Lee, S.Y.; Shen, Y.A.; Wang, L.S.; Chen, Y.J.; Wei, Y.H. High Mitochondrial DNA Copy Number and Bioenergetic Function are Associated with Tumor Invasion of Esophageal Squamous Cell Carcinoma Cell Lines. Int. J. Mol. Sci. 2012, 13, 11228–11246, doi:10.3390/ijms130911228.
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288, doi:10.1073/pnas.0510511103.
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073, doi:10.1038/srep09073.
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264, doi:10.1182/blood-2015-07-655860.
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94, doi:10.1186/1479-5876-11-94.
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429, doi:10.1182/blood.2019001398.
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2018; Volume 137, pp. 37–75. ISBN 978-0-12-815123-5.
- Yan, H.; Guo, B.Y.; Zhang, S. Cancer-associated fibroblasts attenuate Cisplatin-induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem. Biophys. Res. Commun. 2016, 470, 947–954, doi:10.1016/j.bbrc.2016.01.131.
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408, doi:10.1038/srep38408.
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375, doi:10.1038/s41419-019-1581-6.
- Louault, K.; Bonneaud, T.L.; Séveno, C.; Gomez-Bougie, P.; Nguyen, F.; Gautier, F.; Bourgeois, N.; Loussouarn, D.; Kerdraon, O.; Barillé-Nion, S.; et al. Interactions between cancer-associated fibroblasts and tumor cells promote MCL-1 dependency in estrogen receptor-positive breast cancers. Oncogene 2019, 38, 3261–3273, doi:10.1038/s41388-018-0635-z.
- Weigel, K.J.; Jakimenko, A.; Conti, B.A.; Chapman, S.E.; Kaliney, W.J.; Leevy, W.M.; Champion, M.M.; Schafer, Z.T. CAF-Secreted IGFBPs Regulate Breast Cancer Cell Anoikis. Mol. Cancer Res. 2014, 12, 855–866, doi:10.1158/1541-7786.MCR-14-0090.
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M.; et al. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res. 2016, 76, 6495–6506, doi:10.1158/0008-5472.CAN-16-1457.
- Armstrong, T. Type I Collagen Promotes the Malignant Phenotype of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437, doi:10.1158/1078-0432.CCR-03-0825.
- Aoudjit, F.; Vuori, K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 2001, 20, 4995–5004, doi:10.1038/sj.onc.1204554.
- An, J.; Enomoto, A.; Weng, L.; Kato, T.; Iwakoshi, A.; Ushida, K.; Maeda, K.; Ishida-Takagishi, M.; Ishii, G.; Ming, S.; et al. Significance of cancer-associated fibroblasts in the regulation of gene expression in the leading cells of invasive lung cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 379–388, doi:10.1007/s00432-012-1328-6.
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183, doi:10.1038/nature10137.
- Chen, X.; Gu, W.; Wang, Q.; Fu, X.; Wang, Y.; Xu, X.; Wen, Y. C-MYC and BCL-2 mediate YAP-regulated tumorigenesis in OSCC. Oncotarget 2018, 9, 668–679, doi:10.18632/oncotarget.23089.
- Müerköster, S.S.; Werbing, V.; Koch, D.; Sipos, B.; Ammerpohl, O.; Kalthoff, H.; Tsao, M.S.; Fölsch, U.R.; Schäfer, H. Role of myofibroblasts in innate chemoresistance of pancreatic carcinoma-Epigenetic downregulation of caspases. Int. J. Cancer 2008, 123, 1751–1760, doi:10.1002/ijc.23703.
- Wang, L.; Li, X.; Ren, Y.; Geng, H.; Zhang, Q.; Cao, L.; Meng, Z.; Wu, X.; Xu, M.; Xu, K. Cancer‐associated fibroblasts contribute to cisplatin resistance by modulating ANXA 3 in lung cancer cells. Cancer Sci. 2019, 110, 1609–1620, doi:10.1111/cas.13998.
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753, doi:10.15252/emmm.201404346.
- Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125, doi:10.1186/1476-4598-8-125.
- Chen, Z.X.; Pervaiz, S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death Differ. 2007, 14, 1617–1627, doi:10.1038/sj.cdd.4402165.
- Chen, Y.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276, doi:10.1083/jcb.201108059.
- Huang, H.; Shah, K.; Bradbury, N.A.; Li, C.; White, C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014, 5, e1482, doi:10.1038/cddis.2014.419.
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583, doi:10.1038/ncb2488.
- Heiden, M.G.V.; Li, X.X.; Gottleib, E.; Hill, R.B.; Thompson, C.B.; Colombini, M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001, 276, 19414–19419, doi:10.1074/jbc.M101590200.
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233, doi:10.1038/ncb2330.
- Zhang, Z.; Gao, Z.; Rajthala, S.; Sapkota, D.; Dongre, H.; Parajuli, H.; Suliman, S.; Das, R.; Li, L.; Bindoff, L.A.; et al. Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell. Mol. Life Sci. 2020, 77, 1115–1133, doi:10.1007/s00018-019-03209-y.
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520, doi:10.4161/cc.10.15.16585.
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676, doi:10.1038/s41388-019-1014-0.
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140, doi:10.1016/j.cmet.2018.09.012.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598, doi:10.1038/nrc.2016.73.
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777, doi:10.1074/jbc.M112.431973.
- Taddei, M.L.; Giannoni, E.; Raugei, G.; Scacco, S.; Sardanelli, A.M.; Papa, S.; Chiarugi, P. Mitochondrial Oxidative Stress due to Complex I Dysfunction Promotes Fibroblast Activation and Melanoma Cell Invasiveness. J. Signal. Transduct. 2012, 2012, 684592, doi:10.1155/2012/684592.
- Frijhoff, J.; Dagnell, M.; Augsten, M.; Beltrami, E.; Giorgio, M.; Östman, A. The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 2014, 68, 268–277, doi:10.1016/j.freeradbiomed.2013.12.022.
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29, doi:10.1038/s41419-019-2218-5.
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474, doi:10.1164/rccm.201504-0780OC.
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10, doi:10.1038/s41389-017-0011-9.
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485, doi:10.1089/ars.2016.6750.
- Strickaert, A.; Saiselet, M.; Dom, G.; de Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642, doi:10.1038/onc.2016.411.
- Begum, H.M.; Ta, H.P.; Zhou, H.; Ando, Y.; Kang, D.; Nemes, K.; Mariano, C.F.; Hao, J.; Yu, M.; Shen, K. Spatial Regulation of Mitochondrial Heterogeneity by Stromal Confinement in Micropatterned Tumor Models. Sci. Rep. 2019, 9, 11187, doi:10.1038/s41598-019-47593-8.
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120, doi:10.1038/s41586-019-1847-2.
- Yeo, S.K.; Zhu, X.; Okamoto, T.; Hao, M.; Wang, C.; Lu, P.; Lu, L.J.; Guan, J.L. Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. eLife 2020, 9, e58810, doi:10.7554/eLife.58810.
- Choi, J.; Kim, D.H.; Jung, W.H.; Koo, J.S. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res. 2013, 15, R78, doi:10.1186/bcr3472.
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447, doi:10.1242/dmm.029447.
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479, doi:10.1016/j.ccell.2018.01.011.
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056, doi:10.1038/s41467-018-03348-z.
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762, doi:10.1038/s41422-020-0355-0.
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609, doi:10.1038/s41556-018-0083-6.

