 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammed Imad Malki | + 2567 word(s) | 2567 | 2020-10-26 06:51:24 | | | |
| 2 | Bruce Ren | Meta information modification | 2567 | 2020-10-27 04:23:09 | | |
Video Upload Options
DNA damage is well recognized as a critical factor in cancer development and progression. DNA lesions create an abnormal nucleotide or nucleotide fragment, causing a break in one or both chains of the DNA strand. When DNA damage occurs, the possibility of generated mutations increases. Genomic instability is one of the most important factors that lead to cancer development. DNA repair pathways perform the essential role of correcting the DNA lesions that occur from DNA damaging agents or carcinogens, thus maintaining genomic stability. Inefficient DNA repair is a critical driving force behind cancer establishment, progression and evolution. A thorough understanding of DNA repair mechanisms in cancer will allow for better therapeutic intervention.
1. Introduction
DNA damage can alter nucleotide sequences and lead to expression of dysfunctional proteins that impact normal cellular physiology. Sources of DNA damage can be endogenous or exogenous and include reactive oxygen species (ROS) or ionizing radiation [1]. DNA damaging agents can broadly be classified into two different categories: clastogens and aneugens. Clastogens cause chromosomal breaks and induce micronuclei (MN) due to generation of acentric chromosomal fragments. In contrast, aneugens lead to the incorporation of whole chromosomes in MN by generation of aneuploidy that affects cell proliferation and the mitotic spindle apparatus [2].
Genotoxic agents cause structural changes in DNA by disrupting covalent bonds between nucleotides, preventing accurate replication of the genome [3]. Significant numbers of cells in the human body are subjected to DNA damage on a continuous basis which leads to alterations in genome replication and transcription. Although the DNA repair machinery can correct some of these lesions, unrepaired or misrepaired DNA can lead to genome aberrations and mutations that affect cellular function [4]. Genetic defects, especially those occurring in oncogenes, tumor-suppressor genes, genes that control the cell cycle, etc., can impact cell survival or proliferation [5]. Such DNA damage can be carcinogenic [6]. DNA repair proteins trigger checkpoints to recognize sites of damage and either activate corrective pathways or induce apoptosis [7].
Endogenous agents induce replication stress or generate free radicals derived from the oxidative metabolism, whereas exogenous agents such as ionizing or ultraviolet (UV) radiation and chemotherapy induce structural changes such as single strand (SSB) or double strand breaks (DSB) in DNA via base modifications, helix-distorting bulky lesions, or cross-links of DNA strands, and are repaired by biochemically distinct DNA repair pathways [8]. DSBs are the most severe form of DNA damage in eukaryotic cells, because they lead to inefficient repair and cause mutations or induce cell death.
2. Types of DNA Damage
DNA lesions affect a huge number of cells in the human body, occuring at a rate of 10,000 to 1,000,000 molecular lesions per cell per day [9]. Unrepaired or incorrectly repaired DNA damage can lead to serious genome aberrations or mutations, potentially affecting cell survival. However, some mutations change cell proliferation due to defects of certain genes, e.g., oncogene, a tumor-suppressor gene, or a gene that controls the cell cycle.
One of the main sources of DNA damage is ionizing irradiation, which can cause direct or indirect DNA damage leading to changes in the structure of DNA that affects nuclear stability [10]. Ionizing radiation can be of various types such as alpha particles, beta particles or gamma radiation [11]. This radiation releases energy when passing through cellular material and can disrupt proteins and nucleic acids [12]. Irradiation can cause DSB at the phosphodiester backbone of DNA [13]. The level and complexity of DNA damage is influenced by the dose of radiation. Radiation doses can also impact the cellular microenvironment and the type of DNA damage [14]. In addition, other factors play a role in initiating DNA damage, such as reactive oxygen species. Radiation damages cells by direct ionization of DNA and other cellular targets and by indirect effects through ROS [15]. Oxygen-derived free radicals in the tissue environment are produced due to the exposure to ionizing radiation; these include hydroxyl radicals, superoxide anion radicals and hydrogen peroxide. Two-thirds of the damage caused by X-rays and gamma rays are efficient in killing cancer cells. Radiotherapy leads to the production of ROS which affect the survival rate and increase the level of apoptosis in normal cells (Figure 1) [16].
After DNA damage occurs, the DNA repair proteins should identify the site of the damage and determine whether to repair the damage or push the cells towards apoptosis through a DNA damage check point. Apoptosis or programmed cell death (PCD) plays a vital role in maintaining tissue homeostasis by removing diseased or injured cells. Mitochondrial fragmentation within such cells leads to caspase activation and cell death when cells pass through critical checkpoints [17][18]. Conversely, survival pathways such as target of rapamycin complex 1 (TORC1) are activated in response to genotoxic stress to maintain deoxynucleoside triphosphate pools.
Abnormal cell proliferation is one of the hallmarks of cancer [19], but the behavior and the response of cancer cells to the treatments is not well known and still under investigation [20].
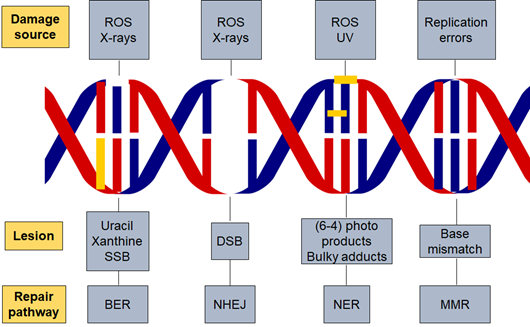
Figure 1. DNA damage and repair pathways. Different factors are responsible for initiating DNA damage such as radiation and reactive oxygen species which cause several types of lesions in the DNA double helix. The repair pathway involved in the process is dependent on the damaging agent and lesion generated. Base excision repair (BER), nucleotide excision repair (NER), non-homologous end joining (NHEJ), reactive oxygen species (ROS) and DNA mismatch repair (MMR).
3. DNA Damage Response
DNA repair pathways are encoded by a class of proteins that detect DNA double stand breaks, chromosomal fragmentation, translocation and deletions, and can correct some alterations [21]. Cells suffer constant and regular insults from genotoxic agents. The DNA damage response (DDR) pathway responds to cellular damage by using signal sensors, transducers and effectors [22]. Such mechanisms help the genome to tolerate or correct damage on an ongoing basis. Endogenous cellular processes produce free radicals, which affect human cells around 10,000 times/day and cause oxidative DNA damage [23]. The presence of DNA damage or DNA replication stress leads to abnormalities in DNA structure which subsequently stimulate the DDR pathway [24].
DDR mechanisms include multiple DNA repair pathways, damage tolerance processes and cell-cycle checkpoints [25]. DNA replication stress activates DDR leading to DNA double-strand breaks (DSBs) and genomic instability [26]. DDR can regulate genomic stability by repairing damaged DNA or removing defective cells by programmed cell death [27]. On the other hand, genomic instability and deregulation of DNA damage repair (DDR) pathways can be associated with cancer progression [28]. Mutations or deletion of genes responsible for regulating cell division or tumor suppressors can also lead to genomic instability and cancer [29]. Genetic alterations that lead to cancer are more likely to occur in actively proliferating tissues. Cells with high rates of proliferation are more susceptible to DNA damage and tumorigenesis [30]. Genomic instability is responsible for tumor progression and the modification of normal cells to cancer cells. In hereditary cancers, the frequency of the mutated base pair is induced due to loss in the function of the DNA repair genes [31][32].
The tumor-suppressor protein Tp53 identifies the presence of DSB and activates the signaling pathways that regulate tumor progression and promote apoptosis. Mutations in the p53 gene affect DNA damage repair and promote cancers [33]. A functional DDR is essential for human health, and dysfunction can lead to several diseases such as immune deficiency, neurodegeneration, premature aging, and cancer. The PIKK kinase family members, ataxia telangiectasia mutated (ATM) and RAD3-related (ATR) are major regulators of DDR. They are sensor proteins and often work together in response to DNA damage signals. ATM and ATR recognize changes in the DNA structure and, as a consequence, mediate downstream protein phosphorylation events and facilitate DDR [34].
Well-studied DNA repair pathways including base excision repair (BER) for SSBs, nucleotide excision repair (NER) for bulky adducts, and non-homologous end joining (NHEJ) and homologous recombination (HR) for DSBs. In addition, there is also DNA mismatch repair (MMR) for the correction of replication errors such as base-pair mismatches and loops/bubbles arising from a series of mismatches [35].
4. Components of the DNA Damage Response
In mammalian cells, the major DDR-signaling components include the protein kinases ATM and ATR, which are induced by DSBs and replication protein A (RPA) that binds to single-stranded DNA (ssDNA) [36][37]. The protein kinases CHK1 and CHK2T are targeted by ATM/ATR. Both are responsible for inhibiting cyclin-dependent kinase (CDK) activity through different mechanisms, which are facilitated by stimulation of the p53 transcription factor [38]. Cell-cycle progression at the G1-S, intra-S and G2-M “cell-cycle checkpoints” are reduced or arrested by the inhibition of CDKs. This step is thought to be essential for improving the chance of DNA repair before replication or mitosis is completed. However, DNA repair is enhanced by ATM/ATR signaling through stimulating DNA repair proteins transcriptionally or post-transcriptionally by modulating their phosphorylation, acetylation or ubiquitylation [39]. The activation of DNA repair proteins acts by recruiting repair factors to the site of damaged DNA. Proteomics studies demonstrate that the DDR regulates additional cellular processes, as this technique recognizes a considerable amount of uncharacterized ATM/ATR-mediated phosphorylation sites [40]. In the event that there is no defect in this mechanism, this will allow efficient DNA repair so that DDR inactivation ensues, which leads to retrieving normal cell functions. On the other hand, if the repairing mechanism is not able to eliminate the damaged DNA, chronic DDR signaling prompts cell apoptosis, causing cell death or a state of stable cell-cycle arrest; both responses function as a potential anti-tumor response [41].
Furthermore, DDR is affected by chromatin structures that may be modified in response to DNA damage [42]. One of the main examples is the phosphorylation of serine-139 of the histone H2A variant. H2AX is mediated by ATM/ATR/DNA-PK, on chromatin located on the sites of the DSB. The stimulation of DSB repair and increased DSB signaling are through the ubiquitin-adduct formation in the DNA damaged regions, and the recruitment of DDR factors besides other chromatin-modifying components [39]. Remarkably, the chromatin relaxation at sites of DSBs is caused due to the activation of ATM [43], and in DDR the H2AX tyrosine-142 phosphorylation is functioning [44][45]. These findings suggest that further investigations are needed on DDR-induced chromatin modifications.
5. DDR and Disease Treatment
The most well-known cancer treatments function based on generating DNA damage, such as radiotherapy and chemotherapy. These types of treatments are efficient, although they cause dose-limited toxicities in normal cells. The rapid proliferation of cancer cells compared with normal cells is due to an impaired DDR. However, the rationale underlying the resistance to cancer therapy is associated with common DNA repair mechanisms. For instance, it has been reported that the treatment of glioblastoma is difficult as a result of the unique properties of their DDR machinery [46]. These findings meditated that using DDR inhibition might promote the efficacy of radiotherapy and DNA-damaging chemotherapies. Moreover, there are many DDR-inhibitor drugs in early stage trials including Mitomycin C, Cisplatin, Etoposide (Topo II), anthracyclines, Epirubicin and Daunorubicin (Topo II) [47][48]. Blocking apoptosis is another potential application for DDR inhibitors to reduce the toxicity levels in normal cells, which are mediated by CHK2 and p53 .
Generally, one or other aspects of the DDR are defected in all cancer cells due to alterations of the behavior of cells during tumor evolution. Therapeutic outcomes are improved when there is a decrease or loss in the DDR factors. There is an exception of poor resistance to the therapeutic effect in the case of disorders in p53 and other pro-apoptotic proteins [49][50]. However, the use of DDR pathway inhibitors has a greater effect on cancer cells than normal cells. In some cases, different DNA repair pathways could be involved and might overlap in function and each pathway might be used as an alternative pathway in repairing DNA damage. An example of the repairing pathway inhibitors includes drugs that target the enzymes that facilitate the repairing process, such as PARP-1, which binds SSBs and BER intermediates. Remarkably, PARP inhibitors are comparatively non-toxic to normal cells, but impact cytotoxicity in homologous recombination deficiency cells, especially in cells which have a defect in BRCA1 or BRCA2 genes [51][52]. The HR-defective cells are defected in BRCA gene and are considered as cancer cells, indicating that the wild-type BRCA allele is completely absent. In patients having one wild-type BRCA allele and one mutant BRCA allele, the HR is unimpaired in their normal cells. HR is required to repair the accumulation of SSBs that are converted later into pernicious DSBs due to inhibition of PARP1. The BRCA1- or BRCA2-deficient cancer cells are not able to repair the lesions in the same way as the normal cells that are repaired by HR [53]. The HR repair is impaired in cancerous cells, and subsequently the tumor cells lead to apoptosis. These observations prove that the defects occur in two different genes or pathways, together resulting in cell apoptosis, whereas defects in one of the two different genes or pathways do not affect the cell survival [54].
In addition, in a phase I trial on PARP inhibitor as a single agent in patients with BRCA mutations using oral PARP inhibitor olaparib in order to prove the safety of olaparib as a single agent, the patients with BRCA-mutated breast, ovarian, or prostate tumors showed a positive response toward this inhibitor [55]. In the later phase II studies, performed on patients with breast or ovarian cancer with germline BRCA mutations, one-third of them had a positive response to the drug with a low level of toxicities [56]. Currently, PARP inhibitors are used to treat BRCA-mutated ovarian cancer and also have been approved for the treatment of advanced BRCA-mutated breast cancer [57]. Further trials using PARP inhibitors on tumors that have HR defects due to mutation or epigenetic inactivation of HR components suggest the applicability of this treatment to be used for ovarian, prostate and pancreatic cancers. Furthermore, the effectiveness of DNA-damaging agents improved after using CHK1 inhibitors, particularly in p53-deficient cells [58].
The discovery of CRISPR/Cas9 technology, which is based on genome editing, could be performed efficiently through targeting the genes that cause cancers and cancer therapy. A CRISPR/Cas9 system was utilized to target the oncogene HER2 leading to inhibition of cell growth in breast cancer cell lines. The addition of PARP inhibitors increased the inhibitory effect [59]. Poly (ADP-ribose) polymerase (PARP) inhibitors are currently used as cancer treatment only in cells defective in the homologous recombination (HR) pathway [60]. The clinical use of PARP inhibitors might extend especially after recognizing the genetic targets that stimulate or mimic HR deficiencies using CRISPR/Cas9. A study demonstrated that TP53 induced glycolysis and apoptosis regulator (TIGAR) is developed in various types of cancers, and the overall survival in ovarian cancer was decreased when the expression of TIGAR was increased [61]. Therefore, in order to improve the sensitivity of cancer cells to olaparib, TIGAR was knocked down which has an impact on metabolic pathways and increased the cytotoxic effects of olaparib. This step causes downregulation of BRCA1 and the Fanconi anemia pathway and promotes programmed cell death of these cells [62].
Improving the methods to distinguish between cancer and normal cells is necessary for the development of diagnostic procedures which help to ameliorate the efficacy of DNA-damaging and DDR-inhibitor therapies. Moreover, screening for DDR-markers as DDR is activated during oncogenesis, is sensitive and beneficial especially for the detection of cancer that might allow efficient detection of pre-malignant disease [63]. Improving therapeutics that stimulate DDR events can be possible in the longer term to control cancer incidence. This experiment was applied to genetically engineered mice expressing p53-dependent DNA damage responses and showed less tumors compared to wild-type mice [64].
References
- Srinivas, U.S.; Tan, B.W.; Vellayappan, B.; Jeyasekharan, A. ROS and the DNA damage response in cancer. Redox Boil. 2019, doi:10.1016/j.redox.2018.101084.
- Terradas, M.; Martin, M.; Tusell, L.; Genescà, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. Mutat. Res. 2010, doi:10.1016/j.mrrev.2010.03.004.
- Cannan, W.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, doi:10.1002/jcp.25048.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, doi:10.1038/nature08467.
- Pucci, B.; Kasten, M.; Giordano, A. Cell Cycle and Apoptosis. Neoplasia 2000, doi:10.1038/sj.neo.7900101.
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, doi:10.3390/ijms19040970.
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; ASM Press: Washington, DC, USA, 2000.
- Arjunan, K.P.; Sharma, V.K.; Ptasinska, S. Effects of Atmospheric Pressure Plasmas on Isolated and Cellular DNA—A Review. Int. J. Mol. Sci. 2015, doi:10.3390/ijms16022971.
- Friedberg, E.C. Fixing Your Damaged and Incorrect Genes; World Scientific: Singapore, Singapore, 2019.
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, doi:10.1016/j.radonc.2013.06.013.
- Abrahamson, S. Adverse Reproductive Outcomes in Families of Atomic Veterans: The Feasibility of Epidemiologic Studies. Radiat. Res. 1995, doi:10.2307/3579267.
- Talty, J.J. Principles of Ionizing Radiation. In Industrial Hygiene Engineering; Elsevier: Amsterdam, The Netherlands, 1998.
- Lomax, M.; Folkes, L.; O’Neill, P. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. 2013, doi:10.1016/j.clon.2013.06.007.
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.-W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, doi:10.3389/fmolb.2014.00024.
- Smith, T.A.; Kirkpatrick, D.R.; Smith, S.; Smith, T.K.; Pearson, T.; Kailasam, A.; Herrmann, K.Z.; Schubert, J.; Agrawal, D.K. Radioprotective agents to prevent cellular damage due to ionizing radiation. J. Transl. Med. 2017, doi:10.1186/s12967-017-1338-x.
- Borek, C. Antioxidants and radiation therapy. J. Nutr. 2004, doi:10.1093/jn/134.11.3207s.
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, doi:10.1016/j.cell.2011.10.033.
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Boil. Cell 2012, doi:10.1091/mbc.E11-11-0926.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, doi:10.1016/s0092-8674(00)81683-9.
- Cooper, G.M.; Hausman, R.E. The Cell: A Molecular Approach, 4th ed.; ASM Press: Washington, DC, USA, 2007.
- Harrison, J.C.; Haber, J.E. Surviving the Breakup: The DNA Damage Checkpoint. Annu. Rev. Genet. 2006, doi:10.1146/annurev.genet.40.051206.105231.
- McGowan, C.H.; Russell, P. The DNA damage response: Sensing and signaling. Curr. Opin. Cell Biol. 2004, doi:10.1016/j.ceb.2004.09.005.
- Blow, J.J.; Lukas, J.; Bartkova, J. DNA Damage Response as an Anti-Cancer Barrier: Damage Threshold and the Concept of ’Conditional Haploinsufficiency’. Cell Cycle 2007, doi:10.4161/cc.6.19.4754.
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Boil. 2013, doi:10.1101/cshperspect.a012716.
- Giglia-Mari, A.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2010, doi:10.1101/cshperspect.a000745.
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, doi:10.1038/nature03485.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, doi:10.1016/j.cell.2011.02.013.
- Bristow, R.G.; Hill, R.P. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, doi:10.1038/nrc2344.
- Fenech, M. Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discov. Today 2002, doi:10.1016/s1359-6446(02)02502-3.
- Bartek, J. DNA damage response, genetic instability and cancer: From mechanistic insights to personalized treatment. Mol. Oncol. 2011, doi:10.1016/j.molonc.2011.07.006.
- Dai, Y.Y.W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, doi:10.4172/2157-2518.1000165.
- Al-Tassan, N.A.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002, doi:10.1038/ng828.
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, doi:10.1126/science.1140735.
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, doi:10.1016/j.molcel.2010.09.019.
- McKinnon, P.J. DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 2009, doi:10.1038/nrn2559.
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Boil. 2008, doi:10.1038/nrm2450.
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, doi:10.1038/nrc1011.
- Riley, T.; Sontag, E.D.; Chen, P.A.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, doi:10.1038/nrm2395.
- Huen, M.S.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res. 2007, doi:10.1038/cr.2007.109.
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, doi:10.1126/science.1140321.
- Campisi, J.; Daddadifagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, doi:10.1038/nrm2233.
- Misteli, T.; Soutoglou, E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009, doi:10.1038/nrm2651.
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nature 2006, doi:10.1038/ncb1446.
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2008, doi:10.1038/nature07668.
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, doi:10.1038/nature07849.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, doi:10.1038/nature05236.
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, doi:10.1038/nrc2342.
- Martin, S.A.; Lord, C.J.; Ashworth, A. DNA repair deficiency as a therapeutic target in cancer. Curr. Opin. Genet. Dev. 2008, doi:10.1016/j.gde.2008.01.016.
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, doi:10.1038/nrm2147.
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genome Res. 2009, doi:10.1101/gad.1815309.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, doi:10.1038/nature03445.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, doi:10.1038/nature03443.
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, doi:10.1111/cas.12366.
- Ashworth, A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008, doi:10.1200/jco.2008.16.0812.
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. N. Engl. J. Med. 2009, doi:10.1056/nejmoa0900212.
- Audeh, M.W.; Carmichael, J.; Penson, R.; Friedlander, M.L.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, doi:10.1016/s0140-6736(10)60893-8.
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, doi:10.1038/nrc2812.
- Chen, Z.; Xiao, Z.; Gu, W.-Z.; Xue, J.; Bui, M.H.; Kovár, P.; Li, G.; Wang, G.; Tao, Z.-F.; Tong, Y.; et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, doi:10.1002/ijc.22198.
- Wang, H.; Sun, W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2017, doi:10.1016/j.canlet.2016.10.033.
- Yap, T.A.; Sandhu, S.; Carden, C.P.; De Bono, J.S. Poly(ADP-Ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, doi:10.3322/caac.20095.
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genome Res. 2020, doi:10.1101/gad.334516.119.
- Fang, P.; De Souza, C.; Minn, K.; Chien, J. Genome-scale CRISPR knockout screen identifies TIGAR as a modifier of PARP inhibitor sensitivity. Commun. Boil. 2019, doi:10.1038/s42003-019-0580-6.
- Aleskandarany, M.A.; Caracappa, D.; Nolan, C.C.; Macmillan, R.D.; Ellis, I.O.; Rakha, E.A.; Green, A.R. DNA damage response markers are differentially expressed in BRCA-mutated breast cancers. Breast Cancer Res. Treat. 2015, doi:10.1007/s10549-015-3306-6.
- Garcia-Cao, I.; Garcia-Cao, M.; Martín-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. ‘Super p53’ mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002, doi:10.1093/emboj/cdf595.


