Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexandra Vladislavovna Sentyabreva | -- | 1867 | 2022-09-28 11:14:27 | | | |
| 2 | Rita Xu | Meta information modification | 1867 | 2022-09-28 11:24:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kosyreva, A.M.; Sentyabreva, A.V.; Tsvetkov, I.S.; Makarova, O.V. Alzheimer’s Disease and Inflammaging. Encyclopedia. Available online: https://encyclopedia.pub/entry/27851 (accessed on 24 July 2026).
Kosyreva AM, Sentyabreva AV, Tsvetkov IS, Makarova OV. Alzheimer’s Disease and Inflammaging. Encyclopedia. Available at: https://encyclopedia.pub/entry/27851. Accessed July 24, 2026.
Kosyreva, Anna Mikhailovna, Alexandra Vladislavovna Sentyabreva, Ivan Sergeevich Tsvetkov, Olga Vasilievna Makarova. "Alzheimer’s Disease and Inflammaging" Encyclopedia, https://encyclopedia.pub/entry/27851 (accessed July 24, 2026).
Kosyreva, A.M., Sentyabreva, A.V., Tsvetkov, I.S., & Makarova, O.V. (2022, September 28). Alzheimer’s Disease and Inflammaging. In Encyclopedia. https://encyclopedia.pub/entry/27851
Kosyreva, Anna Mikhailovna, et al. "Alzheimer’s Disease and Inflammaging." Encyclopedia. Web. 28 September, 2022.
Copy Citation
Alzheimer’s disease is one of the most common age-related neurodegenerative disorders. The main theory of Alzheimer’s disease progress is the amyloid-β cascade hypothesis. One of the factors, which might play a key role in senile plaques and tau fibrils generation due to Alzheimer’s disease, is inflammaging, i.e., systemic chronic low-grade age-related inflammation. The activation of the proinflammatory cell phenotype is observed during aging, which might be one of the pivotal mechanisms for the development of chronic inflammatory diseases, e.g., atherosclerosis, metabolic syndrome, type 2 diabetes mellitus, and Alzheimer’s disease.
inflammation
neurodegeneration
aging
Alzheimer’s disease
1. Introduction
Aging is a complex, dynamic, multistage, and inevitable biological process that leads to a gradual decrease in the adaptive capacity of the body. It is characterized by the development of so-called age-related pathology and an increased probability of death. At the biological level, aging results from various molecular and cellular damage that accumulates over time [1]. The number of elderly and senile people is steadily growing from year to year; therefore, among them, the proportion of patients with age-associated diseases, which are socially significant and increase the healthcare burden, is increasing. These diseases include type 2 diabetes mellitus (T2DM), atherosclerosis, cardiovascular, and neurodegenerative diseases, including the most common type of dementia, which is Alzheimer’s disease (AD). The mechanism of its development is still unclear; however, there is growing data on the relationship of AD with chronic inflammatory diseases, as well as age-associated inflammation, called inflammaging.
2. Aging and Inflammation
The average age of the population of European countries is the highest in the world, and it is increasing. According to WHO forecasts, from 2020 to 2030, the share of people over 60 within the world population will increase by 34%. By 2050 the number of people aged 80 and over will triple, reaching 426 million [1].
Despite the existence of many theories of aging, none of them fully reveals the fundamental mechanisms underlying this complicated process [2]. Theories on aging can be conditionally divided into two main groups: evolutionary and accidental cell damage. The first group includes theories according to which aging is a programmed process that an organism has acquired during evolution. For example, there are theories of antagonistic pleiotropy, adaptive-regulatory, disposable soma, telomeric, immunological, etc. The second combines theories that consider the accumulation of accidental damage as the main cause of aging. Examples of such theories are intoxication, mitochondrial, epigenetic, apoptosis, accumulation of mutations, etc. [3][4].
In the process of aging, 3 types of changes are observed. The primary one is associated with disorders in the genome, such as a high frequency of mutations, shortening of telomeres (Hayflick limit), epigenetic changes, such as methylation and acetylation of DNA sections, and, as a result, a violation of protein homeostasis in cells. The second type of age-associated change is secondary, realized due to the occurrence of primary ones. This group includes mitochondrial dysfunction and cellular senescence [5]. The third type of changes are integrative, such as depletion of the pool of stem cells and disturbances in intercellular interactions [6]. It is customary to distinguish three types of aging—natural or physiological, premature or pathological, and delayed.
Regardless of the initial mechanisms, the aging of the body is accompanied by systemic chronic low-grade inflammation, which in foreign literature is referred to as inflammaging, i.e., inflammation associated with age [7]. Inflammaging was described in all mammalian species, including laboratory rodents, rhesus monkeys [8], and humans [9]. Some authors associate physiological and pathological aging with the hyperproduction of proinflammatory cytokines and inflammatory mediators produced by innate immune cells [10]. Inflammaging is supposed to reduce life expectancy, especially if combined with age-associated diseases such as T2DM, cardiovascular, oncological and neurodegenerative diseases, including AD [11]. In these diseases, concomitant obesity is often observed, against the background of which the severity of inflaming increases [12].
Cell aging is associated with the impossibility of exiting the G1 or G2/M phase of the cell cycle. This process is accompanied by abrupt changes in cell size, shape, and vacuolization of the cytoplasm, as well as functional properties, including an impaired rate of decline, rearrangement of the nucleus and chromatin, and resistance to the apoptosis signal [13]. However, these cells remain functionally active and produce a certain proinflammatory secretion, known is SASP (senescence-associated secretory phenotype), which is characteristic of aging cells. SASPs include proinflammatory cytokines (IL-1α, IL-1β, IL-6, IL-8, TNFα), chemokines (CCL2, CCL5, CCL20), growth factors (TGF, EGF, bFGF, HGF, VEGF), metalloproteinases (MMP-1, -3, -10, -12, -13, -14), extracellular matrix components (fibronectin, collagen, laminin), aging-associated beta-galactosidase (SA-β-Gal), etc. The composition and intensity of SASP expression depend on the cell type, the triggers that led to its activation, and the time elapsed from its onset. With age, the level of expression of the proinflammatory secretome SASP progressively increases, which leads to the accumulation of products of cellular metabolism. These include free radicals, extracellular ATP, nuclear non-histone protein HMGB1 (high-mobility group protein B1), uric acid, products of the impaired metabolism of phospholipids, and cell membrane proteins, which are ceramides, cardiolipin, lipofuscin, and amyloid-β (Aβ), proteins with irregular spatial organization, such as α-synuclein and tau protein, fragments of mitochondrial and nuclear DNA. All this leads to the activation of proinflammatory reactions and the development of low-grade chronic inflammation [14]. An imbalance in the production of pro- and anti-inflammatory cytokines due to cellular aging leads to the formation of a “senile” phenotype and the development of age-associated diseases [15]. Along with proinflammatory reactions in the aging body, the activation of anti-inflammatory mechanisms is observed. According to the literature, compared with young and middle-aged people, older people have an increased level of both proinflammatory cytokines such as IL-1, IL-2, IL-6, IL-8, IL-12, IL-15, IL-17, IL-18, IL-22, IL-23, TNF-α, and IFN-γ, and anti-inflammatory ones like IL-1Ra, IL-4, IL-10, IL-37, and TGF-β1. The imbalance is a probable cause of the development of inflammatory diseases [16].
It was shown that the expression of NF-κB, a nuclear factor that activates the production of proinflammatory cytokines, increases with age in humans and laboratory animals [17]. Compared with mature laboratory rodents, the skin, liver, kidneys, cerebellum, cardiomyocytes, and gastric mucosa NF-κB binding to DNA regions is more stable, which determines its prolonged effect on the expression of target genes [18]. Skin fibroblasts of elderly people and patients with a genetic form of premature aging, known as childhood progeria (Hutchinson-Gilford syndrome), are characterized by a high level of NF-κB activation [19].
With age, the efficiency of eliminating senescent cells by immunocompetent cells decreases, partly due to involutive changes in the immune system. As a result, the number and duration of functioning of SASP-secreting cells increases, ultimately leading to the aging of tissues and the body altogether, and also increases the risk of developing age-related diseases, and AD in particular [20].
Thus, cellular aging is a risk factor for developing age-related diseases, including neurodegenerative ones. There are many theories of aging; however, despite the differences in the initial mechanisms underlying these theories, ultimately, cellular aging is characterized by the activation of the proinflammatory phenotype of SASP-producing cells. A shift in the balance of SASP towards proinflammatory cytokines may be one of the key mechanisms for the development of chronic inflammatory diseases associated with age, such as atherosclerosis, metabolic syndrome, T2DM, and AD.
3. Inflammaging and Alzheimer’s Disease
Inflammation plays a crucial role in the pathogenesis of various neurological disorders and in mediating interactions between the immune and nervous systems, which may be the key to preventing or delaying the onset of most CNS diseases. Local inflammation in the CNS is observed in amyotrophic lateral sclerosis, multiple sclerosis, Parkinson’s disease, and AD [21]. Among the potential mechanisms contributing to chronic inflammation in the CNS in AD, the neuroinflammation hypothesis is dominant. In accordance with this, DAMPs formed during cell damage activate TLR4 on glial cells, which induces inflammatory reactions [22]. Activation of TLRs leads to the assembly of NLRP3, which plays a pivotal role in chronic inflammation in obesity, IR, T2DM, and neurodegenerative diseases. An increase in the level of muramyl dipeptide, which is a component of the bacterial cell wall, was detected in the blood of old mice [23], and a compensatory increase in miR-223 siRNA [24], which inhibits NLRP3 translation [25] in response to developing chronic inflammation, was also observed [26].
DAMPs are formed in the inflammatory process due to caspase-dependent necrotic cell death, known as necroptosis, which may be one of the mechanisms of inflammaging [27]. Royce et al. [26] showed that the level of markers of necroptosis, which is phosphorylated and non-phosphorylated mixed lineage kinase domain, like pseudokinase (MLKL) in white adipose tissue in mice, increases 3-fold with age. Age-related activation of necroptosis markers was accompanied by an increase in the production of IL-6, TNF-α, and IL-1β and several chemokines [26]. According to Caccamo et al. [28], an increase in the expression of necroptosis markers was observed in the brain of AD patients, and the activation of necroptosis in APP/PS1 transgenic mice with AD increased cognitive impairment. Inhibition of necroptosis by necrostatin-1 (a blocker of TNF-1α-dependent necrosis) in APP/PS1 mice resulted in a decrease in the number of amyloid plaques, the production of proinflammatory cytokines TNF-α and IL-1β, and an improvement in cognitive functions [27]. This indicates an association between age-related activation of necroptosis and the development of neurodegenerative diseases, including AD.
One of the processes that also determines the development of inflammaging is autophagy/mitophagy, the destruction of damaged organelles/mitochondria and protein aggregates by the cell, which is impaired with age [29][30]. Accumulations of damaged organelles in cells as a result of inadequate autophagy, as well as mitochondrial DNA formed by mitochondria not subjected to mitophagy, are considered DAMPs and are recognized by PRRs of immunocompetent cells [31][32]. The interaction of DAMPs with PRRs activates the transcription of IL-6, TNF-α, IL-1β, MMP-8, and NLRP3, being key regulators of inflammation [33][34], the increase of which leads to caspase activation and pyroptosis [35].
According to the literature, microglia activation according to the proinflammatory M1 phenotype is observed with aging. It combines with an increase in the production of proinflammatory cytokines by these cells, including IL-1β, TNF-α, and IL-6 [36]. Microglial cells activated via the M1 pathway are found around amyloid plaques and neurons containing neurofibrils in AD [37]. Microglia recognizes Aβ through a number of receptor complexes, including CD14, TLR2, TLR4, α6β1 integrin, CD47, and CD36, and are capable of phagocytizing amyloid [38]. On the one hand, the accumulation of Aβ in the brain of AD patients may be associated with impaired phagocytosis of amyloid by microglia [39]. On the other hand, as a result of inflammaging, prolonged activation of microglia by a proinflammatory phenotype can initiate the formation of Aβ along the amyloid pathway and neurodegeneration [40]. Activation of microglia in AD is combined with mitochondrial dysfunction, decreased oxidative phosphorylation, and the activation of glycolysis [41]. However, despite the huge number of works devoted to the study of the pathogenesis of AD, it has not yet been possible to establish whether glia-associated inflammation in AD is a cause or a consequence of neurodegeneration.
Thus, processes such as cellular aging, leading to senescence of the immune and nervous systems, mitochondrial dysfunction, SASP production, activation of auto- and mitophagy, development of hypoxia, and the activation of HIF1-dependent signaling pathways all play an important role in the development of inflammaging (Figure 1).
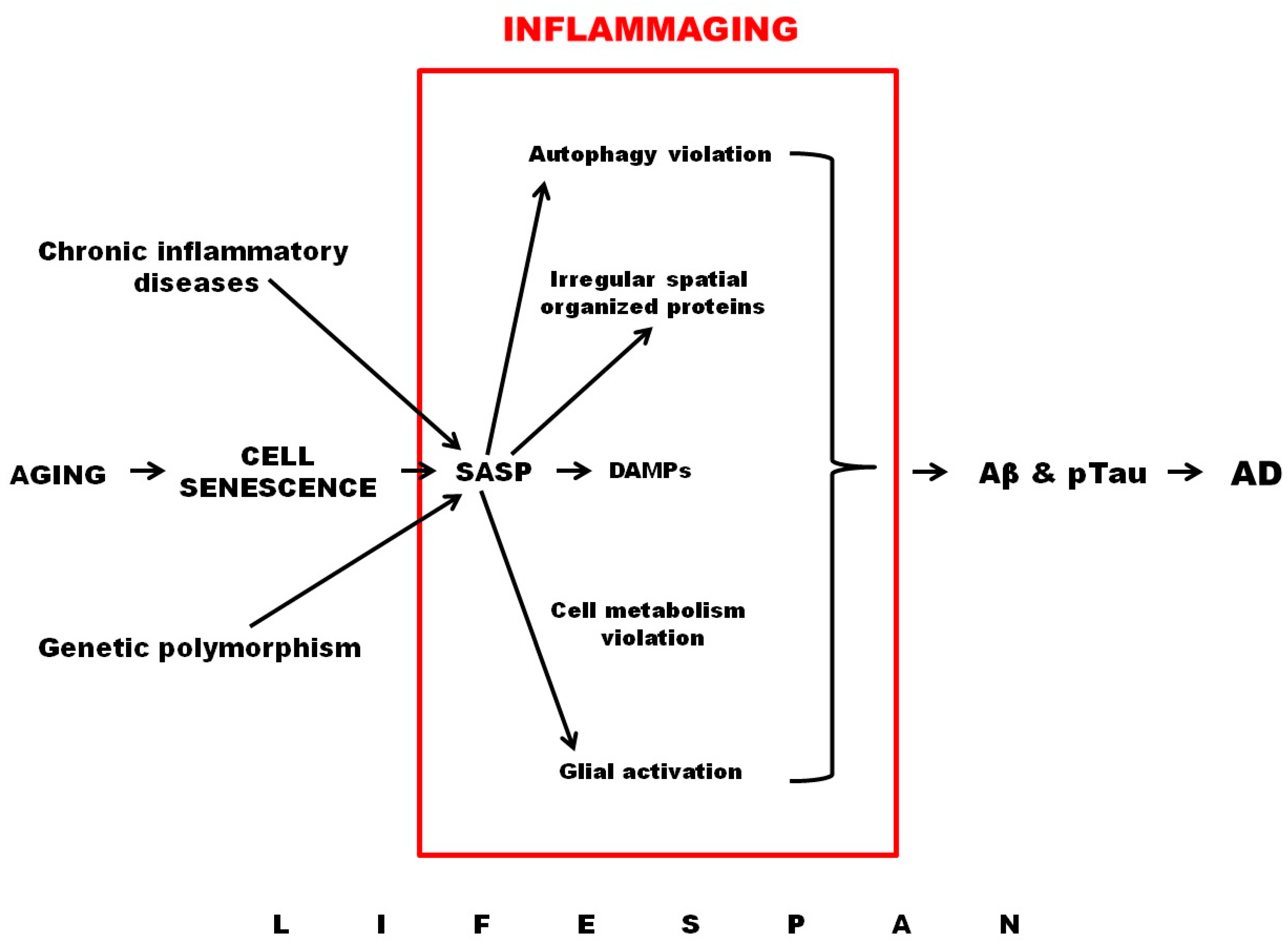
Figure 1. Role of inflammaging in AD development.
References
- World Health Organizatios (WHO). Ageing and Health. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 20 July 2022).
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The Link between Comorbidities, Genetics, and Alzheimer’s Disease. J. Neuroinflamm. 2018, 15, 276.
- Libertini, G.; Graziamaria, C.; Valeria, C.; Olga, S.; Nicola, F. Introduction. In Evolutionary Gerontology and Geriatrics: Why and How We Age; Springer: Cham, Switzerland, 2021; pp. 1–31.
- Skulachev, V.P. New Data on Biochemical Mechanism of Programmed Senescence of Organisms and Antioxidant Defense of Mitochondria. Biochemistry 2009, 74, 1400–1403.
- Ratushnyy, A.Y.; Rudimova, Y.V.; Buravkova, L.B. Replicative Senescence and Expression of Autophagy Genes in Mesenchymal Stromal Cells. Biochemistry 2020, 85, 1169–1177.
- López-Olóriz, J.; López-Cancio, E.; Arenillas, J.F.; Hernández, M.; Jiménez, M.; Dorado, L.; Barrios, M.; Soriano-Raya, J.J.; Miralbell, J.; Cáceres, C.; et al. Asymptomatic Cervicocerebral Atherosclerosis, Intracranial Vascular Resistance and Cognition: The Asia-Neuropsychology Study. Atherosclerosis 2013, 230, 330–335.
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254.
- Didier, E.S.; Sugimoto, C.; Bowers, L.C.; A Khan, I.; Kuroda, M.J. Immune Correlates of Aging in Outdoor-Housed Captive Rhesus Macaques (Macaca mulatta). Immun. Ageing 2012, 9, 25.
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9.
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208.
- Franceschi, C.; Garagnani, P.; Parini, C. Giuliani, and A. Santoro. Inflammaging: A New Immune-Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590.
- Jura, M.; Kozak, L. Obesity and Related Consequences to Ageing. Age 2016, 38, 23.
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420.
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-Aging’. Trends Endocrinol. Metab. 2017, 28, 199–212.
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 030501.
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586.
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. Nrf2 and Nf-κb Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol. 2019, 21, 101059.
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. Nf-Κb Inhibition Delays DNA Damage-Induced Senescence and Aging in Mice. J. Clin. Investig. 2012, 122, 2601–2612.
- Kriete, A.; Mayo, K.L.; Yalamanchili, N.; Beggs, W.; Bender, P.; Kari, C.; Rodeck, U. Cell Autonomous Expression of Inflammatory Genes in Biologically Aged Fibroblasts Associated with Elevated Nf-Kappab Activity. Immun. Ageing 2008, 5, 5.
- Carreno, G.; Guiho, R.; Martinez-Barbera, J.P. Cell Senescence in Neuropathology: A Focus on Neurodegeneration and Tumours. Neuropathol. Appl. Neurobiol. 2021, 47, 359–378.
- Trovato, A.; Siracusa, R.; Di Paola, R.; Scuto, M.; Ontario, M.L.; Bua, O.; Di Mauro, P.; Toscano, M.A.; Petralia, C.C.T.; Maiolino, L.; et al. Redox Modulation of Cellular Stress Response and Lipoxin A4 Expression by Hericium Erinaceus in Rat Brain: Relevance to Alzheimer’s Disease Pathogenesis. Immun. Ageing 2016, 13, 23.
- Pang, Y.; Fan, L.-W. Dysregulation of Neurogenesis by Neuroinflammation: Key Differences in Neurodevelopmental and Neurological Disorders. Neural Regen. Res. 2017, 12, 366–371.
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466.
- Gombar, S.; Jung, H.J.; Dong, F.; Calder, B.; Atzmon, G.; Barzilai, N.; Tian, X.-L.; Pothof, J.; Hoeijmakers, J.H.; Campisi, J.; et al. Comprehensive Microrna Profiling in B-Cells of Human Centenarians by Massively Parallel Sequencing. BMC Genom. 2012, 13, 353.
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: Nf-Kappab Activating Pattern Recognition and Cytokine Receptors License Nlrp3 Inflammasome Activation by Regulating Nlrp3 Expression. J. Immunol. 2009, 183, 787–791.
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-Del-Olmo, I.; Nichols, E.-M.; Davis, D.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for Nlrp3 Inflammasome Activation in Human Monocytes in Vitro. Front. Immunol. 2020, 11, 565924.
- Royce, G.H.; Brown-Borg, H.M.; Deepa, S.S. The Potential Role of Necroptosis in Inflammaging and Aging. Geroscience 2019, 41, 795–811.
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis Activation in Alzheimer’s Disease. Nat. Neurosci. 2017, 20, 1236–1246.
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective Autophagy of Intracellular Organelles: Recent Research Advances. Theranostics 2021, 11, 222–256.
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. Ripk1 Mediates a Disease-Associated Microglial Response in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797.
- Green, D.R.; Levine, B. To Be or Not to Be? How Selective Autophagy and Cell Death Govern Cell Fate. Cell 2014, 157, 65–75.
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy Modulation, a New Player in the Race against Als. Int. J. Mol. Sci. 2022, 22, 740.
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. Damps as Mediators of Sterile Inflammation in Aging-Related Pathologies. Ageing Res. Rev. 2015, 24, 29–39.
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the Regulation of Innate Immune Responses. Protein Cell 2016, 7, 11–16.
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An Overview of the Non-Canonical Inflammasome. Mol. Asp. Med. 2020, 76, 100924.
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘on’ and ‘Off’ Signals Control Microglia. Trends Neurosci. 2007, 30, 596–602.
- Gui, Y.; Marks, J.D.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Characterization of the 18 Kda Translocator Protein (Tspo) Expression in Post-Mortem Normal and Alzheimer’s Disease Brains. Brain Pathol. 2020, 30, 151–164.
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Mitochondrially-Targeted Therapeutic Strategies for Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 753–771.
- Holthoff, V.A.; Ferris, S.; Ihl, R.; Robert, P.; Winblad, B.; Gauthier, S.; Sternberg, K.; Tennigkeit, F. Validation of the Relevant Outcome Scale for Alzheimer’s Disease: A Novel Multidomain Assessment for Daily Medical Practice. Alzheimers Res. Ther. 2011, 3, 27.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472.
- Fairley, L.H.; Wong, J.H.; Barron, A.M. Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Disease. Front. Immunol. 2021, 12, 624538.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
950
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
28 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No