Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yumin Luo | -- | 2636 | 2022-09-26 16:54:52 | | | |
| 2 | Sirius Huang | + 2 word(s) | 2638 | 2022-09-27 03:08:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hu, Y.; Luo, Y.; Zheng, Y. Nrf2 Pathway in Ischemic Cerebral Vascular Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/27607 (accessed on 09 August 2026).
Hu Y, Luo Y, Zheng Y. Nrf2 Pathway in Ischemic Cerebral Vascular Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/27607. Accessed August 09, 2026.
Hu, Yue, Yumin Luo, Yangmin Zheng. "Nrf2 Pathway in Ischemic Cerebral Vascular Diseases" Encyclopedia, https://encyclopedia.pub/entry/27607 (accessed August 09, 2026).
Hu, Y., Luo, Y., & Zheng, Y. (2022, September 26). Nrf2 Pathway in Ischemic Cerebral Vascular Diseases. In Encyclopedia. https://encyclopedia.pub/entry/27607
Hu, Yue, et al. "Nrf2 Pathway in Ischemic Cerebral Vascular Diseases." Encyclopedia. Web. 26 September, 2022.
Copy Citation
Cerebrovascular disease is highly prevalent and has a complex etiology and variable pathophysiological activities. It thus poses a serious threat to human life and health. Many studies have found some effects of oxidative stress and autophagy on cerebrovascular diseases. Nuclear factor erythroid 2-related factor 2 (Nrf2) molecules, which are closely associated with oxidative stress, are discussed.
ischemic cerebral vascular diseases
oxidative stress
Nrf2
1. Introduction
Globally, stroke is the second leading cause of death among people over the age of 60 years, and also the leading cause of permanent disability, characterized by high morbidity, disability, mortality, and recurrence rates [1][2]. Generally, stroke occurs as two types: ischemic and hemorrhagic. Ischemic stroke is the main type of stroke and the third leading cause of disability worldwide [3]. Approximately 80–85% of stroke events occur due to thrombogenesis or embolism in the cerebral artery, which induces cerebral ischemia, resulting in an insufficient blood supply in the ischemic area, especially in the middle cerebral artery [4]. Owing to the complexity of the causes, the diversity of pathophysiological changes, and the instability of disease changes after the onset of cerebrovascular diseases, current treatment strategies are imperfect; thus, side effects often occur. Currently, recombinant tissue plasminogen activator (rtPA) thrombolysis and arterial mechanical thrombectomy are clinically applied for the treatment of ischemic stroke. However, there is a strict treatment time window (4.5 h after stroke onset) for thrombolysis, which makes rtPA thrombolysis suitable for only approximately 5% of stroke patients [5]. In addition, this therapy can induce serious side effects, such as blood-brain barrier (BBB) damage and hemorrhage transformation [6]. Therefore, it is necessary to explore new effective clinical treatments with less toxicity and fewer side effects.
Drugs with significant therapeutic effects on ischemic stroke have become a hot topic in current research, especially active ingredients extracted from natural sources, because of their few associated side effects [7]. However, studies have mainly targeted the pathophysiological changes of stroke patients. The glutamate excitatory toxicity increases due to the decreased blood supply in ischemic areas with an insufficient energy and oxygen supply after an ischemic stroke. With an increase in the excitatory toxicity, the intracellular concentrations of sodium and calcium ions, along with the extracellular concentrations of potassium ions increase. Intracellular calcium overload leads to an increased release of free radicals and NO, resulting in mitochondrial dysfunction and DNA damage, thus inducing various pathophysiological changes, such as oxidative stress, autophagy hyperactivation, inflammatory response, neuronal apoptosis, and vascular damage [8]. Various pathophysiological activities are activated successively or simultaneously, forming a complex network. Diverse pathways and molecular interactions complicate the pathophysiological activities associated with stroke. For instance, oxidative stress and autophagy play important roles in pathological changes.
Oxidative stress is caused by an imbalance in the oxidant and antioxidant activities, generating an excess of reactive oxygen species (ROS), which oxidize macromolecular substances, resulting in attendant cellular damage [9]. Oxidants participate in cell death/survival signaling pathways and mediate mitochondrial dysfunction, resulting in reduced neuronal cell survival and tissue damage after cerebral ischemia [10]. Multiple in vivo antioxidant mechanisms activate to prevent the damage caused by oxidative stress after its initiation, and the main resistance system involves antioxidant enzymes. These enzymes include the erythrocyte antioxidant enzymes superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase (CAT) [11]. In addition, the expression of antioxidant genes increases, among which Nrf2 plays a key role as a transcription factor that initiates the expression of antioxidant genes. The following section details the structure and Nrf2- involved in ischemic cerebrovascular diseases.
2. Nrf2 Pathway and Ischemic Cerebrovascular Diseases
2.1. Structure of Nrf2
Nrf2 is a member of the Cap-n-collar (CNC) family of basic leucine zipper (bZIP) proteins and is an activator of β-globulin genes. Nrf2 is a 66 kDa protein containing 605 amino acid residues and seven highly conserved domains, the Nrf2-ECH homology (Neh) domains, in humans [12]. These seven domains are involved in regulating structural stability and transcriptional activity. The N-terminal domain of Nrf2 is the Neh2 domain, which is responsible for regulating the interactions between Nrf2 and Kelch-like-ECH-associated protein 1 (Keap1), a Nrf2 inhibitor, at nanomolar concentrations (KD ~5 nM), as well as the stability of Nrf2, and the degradation of ubiquitin. The Neh2 domain interacts with Keap1 through two motifs: high-affinity ETGE (KD ~5 nM) and low-affinity DLG (KD ~1 μM). Both motifs are essential for Keap1 regulation [13][14][15]. The Neh1 domain contains a basic leucine zipper module that acts as a heterodimer with the transcription partner small Maf proteins (sMafs) and binds DNA, allowing Nrf2 to bind to the antioxidant response element (ARE) sequence. In addition, the Neh1 domain can bind to UbcM2, a ubiquitin ligase of E2, to regulate Nrf2 protein stability. Upon the release of Neh1 from Keap1, the nuclear localization signal necessary for the nuclear translocation of Nrf2 is exposed [16]. The C-terminal domain of Nrf2 is the Neh3 domain, which interacts with the transcription coactivator CHD6 (a chromo-ATPase/helicase DNA-binding protein) and is responsible for the transactivation of ARE-dependent genes after nuclear chromatin remodeling [17]. Neh3 is a trans-activator domain, and the deletion of the Neh3 domain causes Nrf2 to lose its ability to bind ARE for gene expression, maintaining intact dimers, DNA binding, and subcellular localization [17]. Neh4 and Neh5 are transcriptional activation domains that bind to coactivator cyclic adenosine monophosphate response element-binding proteins to activate transcription [18][19]. The Neh5 domain also regulates Nrf2 cellular localization, while Neh6 regulates the Keap1-dependent degradation of Nrf2 and represents a binding platform for the β-transducin repeat-containing protein (β-TrCP) [16]. β-TrCP is a substrate adaptor for the S-phase kinase-associated protein 1 (SKP1)–Cul1–RING-box protein (Rbx1)/Roc1 ubiquitin ligase complex The Neh6 domain negatively regulates Nrf2 through DSGIS and DSAPGS motifs. DSGIS motif increases the ability of β-TrCP to ubiquitinate Nrf2 and promotes its rapid conversion [20][21]. Neh7 binds to the nuclear receptor–retinoic X receptor alpha (RXRα), which inhibits Nrf2 transcription [22]. The structure of Nrf2 is shown in Figure 1A.
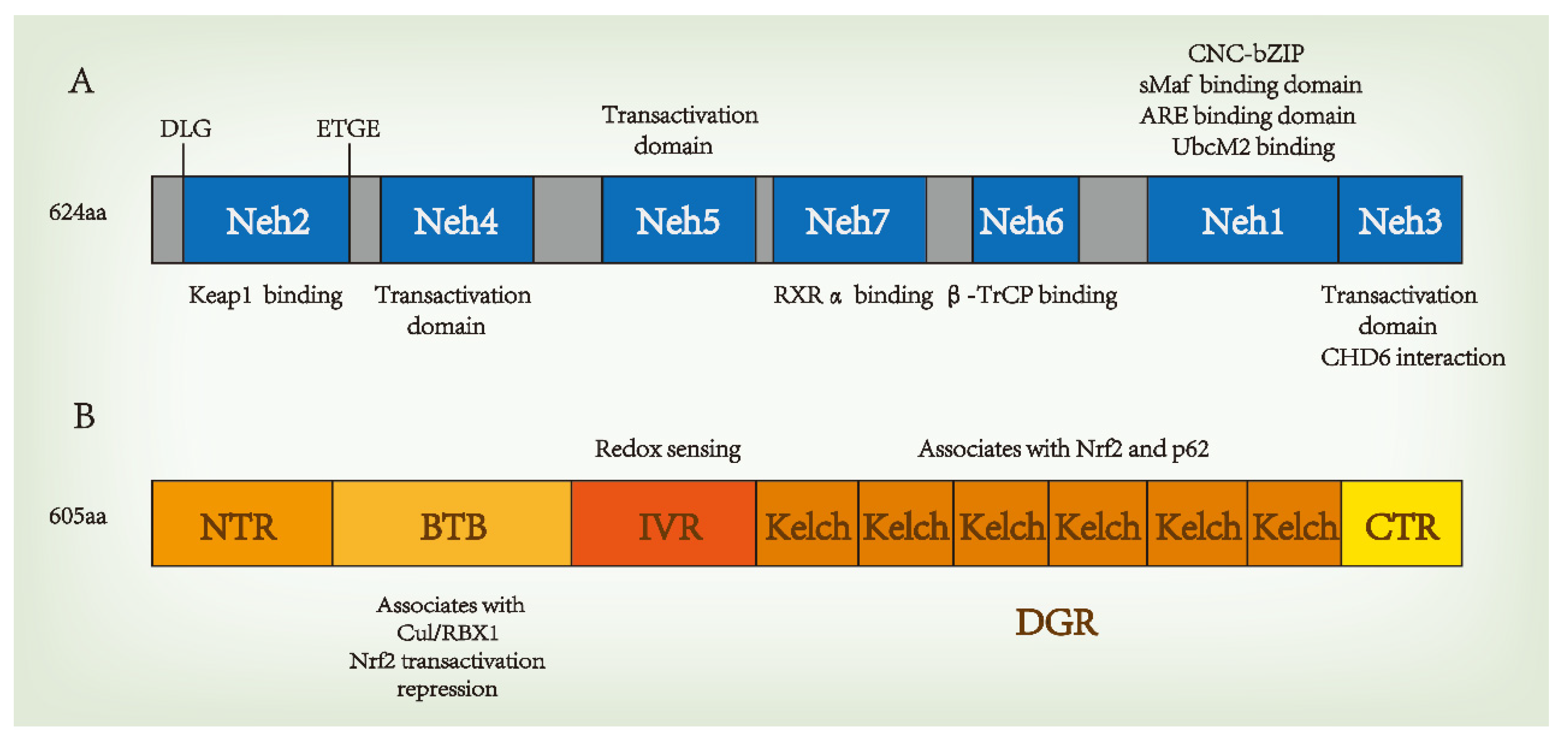
Figure 1. Domain structures of nuclear factor erythroid 2-related factor 2 (Nrf2) (A) and Kelch-like-ECH-associated protein 1 (Keap1) (B). β-TrCP, a β-transducin repeat-containing protein. CNC, cap‘n’collar; bZIP, basic region leucine zipper; sMafs, musculoaponeurotic fibrosarcoma protein; Cul3, Cullin 3; RXRα, retinoic X receptor alpha.
Keap1 is a substrate adaptor for Cul3-containing E3 ubiquitin ligase, which is the main intracellular regulatory factor and cytoplasmic inhibitory protein of Nrf2. Keap1 has five domains, including the N-terminal region (NTR), the Broad-complex, Tramtrack and Bric-à-Brac (BTB) domain, the intervening region (IVR), the Kelch/double glycine repeat (DGR) domain, and the C-terminal region (CTR). Each of these plays an important role in inhibiting Nrf2 activity. The BTB is a homodimerization domain that sequesters Nrf2 in the cytoplasm and represses Nrf2 transactivation [23]. Keap1 binds Cul3 through its N-terminal BTB and binds to the Neh2 domain of Nrf2 through the C-terminal Kelch domain, thus bridging Cul3 and Nrf2 to regulate the degradation of Nrf2 [24]. The IVR domain contains active cysteine residues that act as redox sensors, and C273A and C288A are known to be necessary for Keap1 to inhibit Nrf2 activation [25]. The DGR domain contains six Kelch repeats, which are related to the Neh2 domain of Nrf2 bound to Keap1, and associates with p62 [26]. The structure of Keap1 is shown in Figure 1B.
Keap1 is a part of the Cul3-containing E3 ubiquitin ligase and strongly regulates the activity of the transcription factor Nrf2 through targeted ubiquitination and proteasome-dependent degradation. In homeostasis, two Keap1 molecules interact with one Nrf2 molecule through its DLG and ETGE motifs, thus promoting the ubiquitination and degradation of Nrf2. In the Nrf2-Keap1 complex, DLG functions as a latch, and ETGE functions as a hinge. The “hinge and latch” model forms the stress sensing mechanism, wherein DLG with a low affinity with Nrf2 acts as a latch to turn the degradation of Nrf2 on or off via ubiquitination. When oxidative stress and other stress factors occur, DLG dissociates from Keap1, and Nrf2 separates from the Nrf2-Keap1 complex, translocating into the nucleus, interacting with transcription partner sMafs and binding to ARE to begin transcription activity. This is the classic regulation of Nrf2 transcription. Advances in research have found that Keap1 has a variety of stress receptors and inactivation modes, from oxidative stress and cell metabolism to autophagy disorder, and the regulation of Nrf2 activity from multiple cellular inputs. In mammals, the Nrf2-Keap1 system plays a pivotal role in resisting stress and maintaining homeostasis. The molecular regulatory mechanism of Nrf2 is shown in Figure 2.
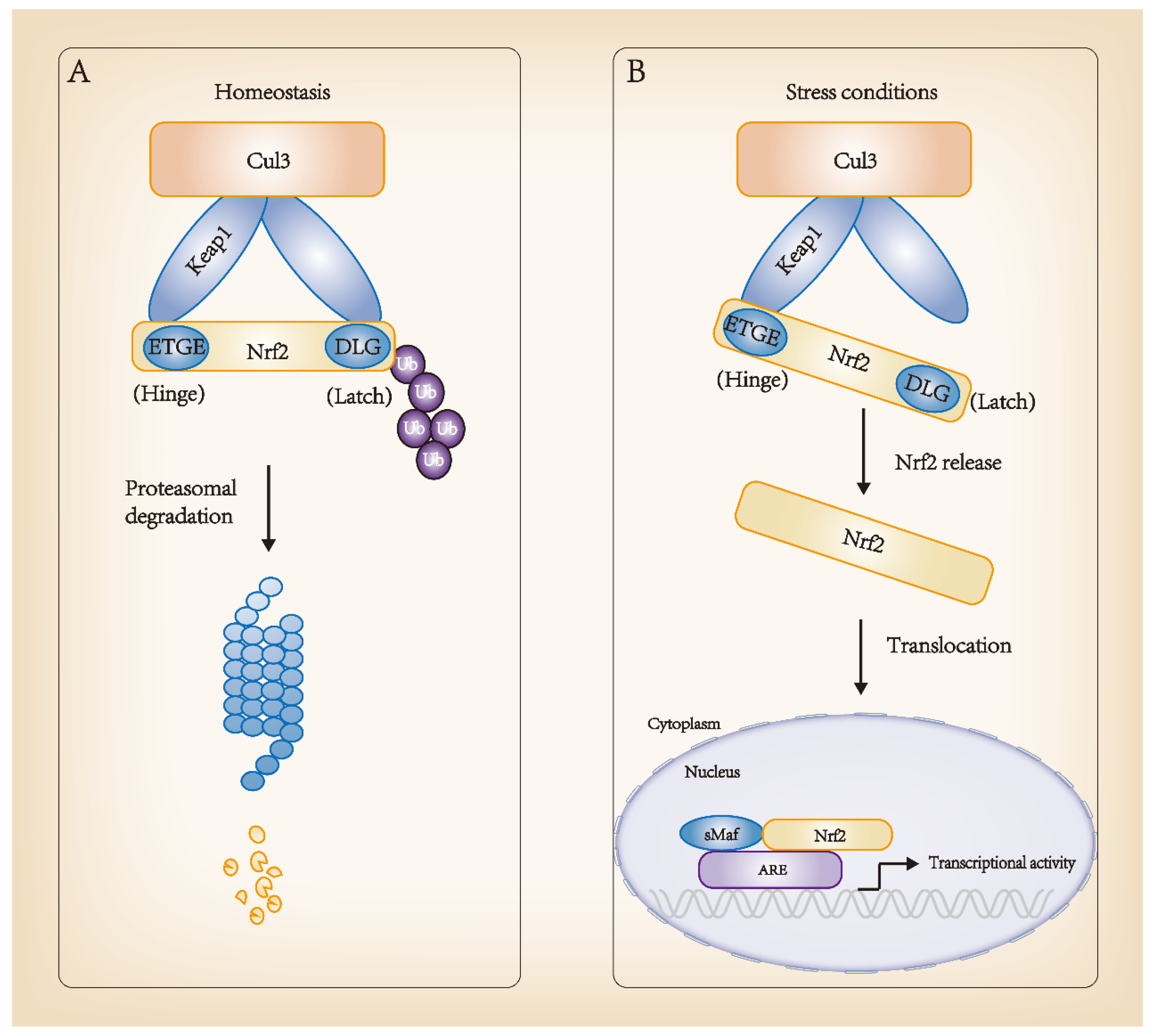
Figure 2. Regulation of Nrf2 molecular mechanisms. (A) Homeostasis: high-affinity ETGE and low-affinity DLG sites of Nrf2 bind to its inhibitor, Keap1, in the cytoplasm and interact with Cul3-Rbx1 E3 ubiquitin ligase, consistently leading to Nrf2 ubiquitination and proteasomal degradation. (B) Stress conditions: Keap1 changes its conformation and dissociates from Nrf2. Nrf2 translocates into the nucleus and forms a dimer with sMaf. The complex binds to ARE sequences, promoting the transcription of target genes, such as detoxification genes. ARE, antioxidant responsive element.
2.2. Nrf2-Involved in Ischemic Cerebrovascular Diseases
Based on different types of animal models of cerebral ischemia, numerous studies have reported the dynamic regulation of Nrf2 after cerebral ischemia. The cerebral ischemia animal models used in these studies included permanent cerebral ischemia models (pMCAO), transient cerebral ischemia models (tMCAO) and global cerebral ischemia models. In most pMCAO and tMCAO animals, the expression of the Nrf2 protein, its downstream antioxidant target genes and its expressed target proteins (heme oxygenase1 (HO-1), NAD(P)H: Quinone oxidoreductase I (NQO1), etc.) were upregulated in different ischemic sites (cortex, hippocampus, etc.) and different ischemic cells (neurons, astrocytes, microglia, etc.) after cerebral ischemia [27][28][29][30][31]. However, a few studies did not find a statistically significant increase in Nrf2 expression [32], or even a decrease in Nrf2 mRNA expression [33], thus indicating the need for further exploration. In addition, studies on global cerebral ischemia models seem to be controversial owing to the influence of many factors, such as animal background, age, difference in ischemic time, and observations at different ischemic sites [34].
Nrf2 has been found to play a protective role after cerebral ischemia in several Nrf2 knockout animal models [35][36]. In several pMCAO experiments, severe cortical infarct volumes and neurological deficits were found in Nrf2 knockout animals [37][38][39]. Therefore, studies on experimental animal models of cerebral ischemia revealed that Nrf2 is a potential target for intervention in ischemic stroke. Nrf2 is expected to stimulate cellular protective responses against harmful ischemic events. At present, several candidate Nrf2 inducers have been identified in Nrf2-knockdown cerebral ischemia animal models that may play a protective role in the brain. These include dimethyl fumarate (DMF), monomethyl fumarate, sulforaphane (SFN), and tert-butylhydroquinone [39][40][41]. However, some studies have shown distinct results regarding the role of Nrf2 in cerebral ischemia, warranting further investigation. There are multiple possible reasons for the contrary results: selection differences for Nrf2 antibodies, different types of Nrf2-related cells that play a role in neuroprotection, different Nrf2 knockout lines, whether the expression of protective Nrf2-activated enzymes is sufficiently high, whether the Nrf2 activator can pass through the BBB and directly activate Nrf2, differences in detected ischemic brain tissue sites, and differences in detected ischemia time points and markers. Owing to these factors, overall differences may arise in the experimental results. Hence, further rigorous and in-depth studies are necessary to provide information for Nrf2-targeted interventions and realize their prospects for clinical transformation and application [34].
Ischemic brain tissues are damaged by increased aseptic inflammation and oxidative stress caused by ROS, which results from the resupply of blood flow after cerebral ischemia-reperfusion (I/R). ROS can cause membrane lipid peroxidation and destroy the membrane integrity, leading to protein degeneration, DNA damage, and cell death. Consequently, ROS-stimulated oxidative stress is an important pathological change associated with cerebral I/R injury. It has been found that Nrf2 confronts oxidative stress through multiple signaling pathways. Several studies have found that the Nrf2-Keap1-ARE signaling pathway plays an anti-oxidative stress role in cerebral I/R injury [42]. Nrf2 activation acts as a transcription factor against oxidative stress and regulates the expression of antioxidant enzymes that act as ROS scavengers and electrophilic neutralizers. These enzymes include catalase, superoxide dismutase, thioredoxin, thioredoxin reductase, glutathione peroxidase, glutathione reductase, sulfiredoxin, NQO1, HO-1, glutaredoxin, and glutathione S-transferase [43][44][45]. Moreover, Nrf2 interacts with the NF-κB signaling pathway to regulate redox homeostasis and cellular responses to stress and inflammation [46]. Multiple signals activate NF-κB by degrading the IκB proteasome and subsequently translocating to the nucleus to induce the expression of inflammatory factors and anti-oxidative stress genes, thereby regulating cellular redox homeostasis, stress, and inflammatory responses. Furthermore, multiple studies have found that excessive ROS activates Nrf2, regulates the transcription and expression of the p62 gene, and participates in the protein degradation regulated by autophagy after cerebral ischemia [47]. Therefore, the ROS-Nrf2-p62 signaling pathway regulates oxidative stress and autophagy after cerebral ischemia and plays a protective role in the cells. Glycogen synthase kinase 3β (GSK-3β) is an inhibitor of Nrf2, which functions by inhibiting nuclear translocation and degrades Nrf2 by phosphorylation [20]. This process occurs in a Keap1-independent manner. However, GSK-3β is activated by hydrogen peroxide; therefore, the regulation of Nrf2 by GSK-3β may also occur through oxidative stress sensitivity. Recent studies have focused on modulating Nrf2 by GSK-3β to counter the altered oxidative environment of brain tissues after cerebral ischemia. However, previous studies in some animal models have shown the opposite results regarding the possibility of GSK-3β regulating Nrf2 and protecting against cerebral ischemia, while some existing studies have defects in their research methods. Therefore, whether GSK-3β inhibition can enhance Nrf2 activity and play a protective role after cerebral ischemia requires further exploration and confirmation [48].
The increased permeability of the BBB and impairment of neurological function will occur after an ischemic stroke. The Nrf2 activator SFN alleviates the blood–brain barrier injury and inhibits disease progression by activating Nrf2 and HO-1 in perivascular astrocytes around infarction, thus alleviating neurological impairment. Consequently, the activation of the Nrf2-HO-1 signaling pathway may play a neuroprotective role after cerebral ischemia [40]. Other studies have found that lycopene pretreatment of the bilateral common carotid artery occlusion (BCCAO) mouse model for 7 days can reduce neuronal apoptosis and improve neurological function scores by activating the Nrf2/HO-1 pathway [49]. Additionally, DMF induces the Nrf2/HO-1 pathway. Using a rat MCAO model, Yang et al. found that DMF significantly reduced the volume of cerebral infarction, alleviated cerebral edema, decreased cell death, and improved neurological defects [39]. In a study on the neuroprotective effect of andrographolide on stroke, Yang et al. found that a p38 mitogen-activated protein kinase (MAPK) inhibitor significantly inhibited Nrf2 phosphorylation and reduced the expression of the Nrf2 downstream target gene, HO-1, suggesting that p38MAPK may be an upstream regulatory factor for Nrf2 activation. In summary, andrographolide plays a neuroprotective role by activating the p38MAPK/Nrf2/HO-1 signaling pathway [50].
The Nrf2 signaling pathway can not only combat oxidative stress, but also promotes neovascularization. Huang et al. found that under hypoxia, Nrf2-knockout brain microvascular endothelial cells can inhibit VEGF expression through the PI3K/AKT pathway, thereby inhibiting angiogenesis [51]. Data indicated that hypoxia induces a transient increase in Nrf2, and that Nrf2 plays an important role in cerebral microangiogenesis. In hypoxia-induced bEnd.3 cells, Nrf2 may regulate the proliferation and migration of vascular endothelial cells and the formation of tubular structures through the PI3K/AKT pathway [51]. In addition, Bai et al. found in a tMCAO rat model that epigallocatechin-3-gallate, the active ingredient in green tea, can promote angiogenesis and reduce oxidative stress by activating the MAPK/Nrf2/VEGF signaling pathway [52].
In addition to its critical role in ischemic stroke, Nrf2 is also involved in other ischemic cerebrovascular diseases. Endothelial cell injury is an early pathological change in cerebral small vessel disease, and oxidative stress is the key factor in this process. Increased Nrf2 expression can significantly improve endothelial cell injury by reducing oxidative stress [53]. Mixed dementia presents with mutually reinforcing neurodegeneration and vasculopathy, with all the features of Alzheimer’s disease (AD) and vascular dementia. The enhancement of cAMP and/or cGMP signaling has been found to exert beneficial effects via Nrf2 activation [54].
References
- Ajoolabady, A.; Wang, S.; Kroemer, G.; Penninger, J.M.; Uversky, V.N.; Pratico, D.; Henninger, N.; Reiter, R.J.; Bruno, A.; Joshipura, K.; et al. Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics. Pharmacol. Ther. 2021, 225, 107848.
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464.
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603.
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788.
- Reeves, M.J.; Arora, S.; Broderick, J.P.; Frankel, M.; Heinrich, J.P.; Hickenbottom, S.; Karp, H.; LaBresh, K.A.; Malarcher, A.; Mensah, G.; et al. Acute stroke care in the US: Results from 4 pilot prototypes of the Paul Coverdell National Acute Stroke Registry. Stroke 2005, 36, 1232–1240.
- Hu, Y.; Zheng, Y.; Wang, T.; Jiao, L.; Luo, Y. VEGF, a Key Factor for Blood Brain Barrier Injury After Cerebral Ischemic Stroke. Aging Dis. 2022, 13, 647–654.
- Dong, Q.; Lin, X.; Shen, L.; Feng, Y. The protective effect of herbal polysaccharides on ischemia-reperfusion injury. Int. J. Biol. Macromol. 2016, 92, 431–440.
- Meng, H.; Jin, W.; Yu, L.; Xu, S.; Wan, H.; He, Y. Protective effects of polysaccharides on cerebral ischemia: A mini-review of the mechanisms. Int. J. Biol. Macromol. 2021, 169, 463–472.
- Sachdev, S.; Ansari, S.A.; Ansari, M.I.; Fujita, M.; Hasanuzzaman, M. Abiotic Stress and Reactive Oxygen Species: Generation, Signaling, and Defense Mechanisms. Antioxidants 2021, 10, 277.
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517.
- Fabian, E.; Bogner, M.; Elmadfa, I. Age-related modification of antioxidant enzyme activities in relation to cardiovascular risk factors. Eur. J. Clin. Investig. 2012, 42, 42–48.
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745.
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075.
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846.
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617.
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733.
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906.
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868.
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527.
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781.
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133.
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108.
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552.
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953.
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770.
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107.
- Miao, Z.Y.; Xia, X.; Che, L.; Song, Y.T. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of Nrf2 expression in ovariectomized rats. Neurol. Res. 2018, 40, 689–695.
- Cai, M.; Guo, Y.; Wang, S.; Wei, H.; Sun, S.; Zhao, G.; Dong, H. Tanshinone IIA Elicits Neuroprotective Effect Through Activating the Nuclear Factor Erythroid 2-Related Factor-Dependent Antioxidant Response. Rejuvenation Res. 2017, 20, 286–297.
- An, P.; Wu, T.; Yu, H.; Fang, K.; Ren, Z.; Tang, M. Hispidulin Protects Against Focal Cerebral Ischemia Reperfusion Injury in Rats. J. Mol. Neurosci. 2018, 65, 203–212.
- Zhao, Y.; Fu, B.; Zhang, X.; Zhao, T.; Chen, L.; Zhang, J.; Wang, X. Paeonol pretreatment attenuates cerebral ischemic injury via upregulating expression of pAkt, Nrf2, HO-1 and ameliorating BBB permeability in mice. Brain Res. Bull. 2014, 109, 61–67.
- Ya, B.L.; Li, H.F.; Wang, H.Y.; Wu, F.; Xin, Q.; Cheng, H.J.; Li, W.J.; Lin, N.; Ba, Z.H.; Zhang, R.J.; et al. 5-HMF attenuates striatum oxidative damage via Nrf2/ARE signaling pathway following transient global cerebral ischemia. Cell Stress Chaperones 2017, 22, 55–65.
- Zhang, W.; Song, J.K.; Yan, R.; Li, L.; Xiao, Z.Y.; Zhou, W.X.; Wang, Z.Z.; Xiao, W.; Du, G.H. Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol. Sin. 2018, 39, 1259–1272.
- Clausen, B.H.; Lundberg, L.; Yli-Karjanmaa, M.; Martin, N.A.; Svensson, M.; Alfsen, M.Z.; Flaeng, S.B.; Lyngso, K.; Boza-Serrano, A.; Nielsen, H.H.; et al. Fumarate decreases edema volume and improves functional outcome after experimental stroke. Exp. Neurol. 2017, 295, 144–154.
- Liu, L.; Locascio, L.M.; Dore, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 2019, 10, 153.
- Dore, S. Neuroprotective effert of carbon monoxide and Nrf2 in cerebral ischemia. Springerplus 2015, 4, L44.
- Liu, L.; Vollmer, M.K.; Fernandez, V.M.; Dweik, Y.; Kim, H.; Dore, S. Korean Red Ginseng Pretreatment Protects Against Long-Term Sensorimotor Deficits After Ischemic Stroke Likely Through Nrf2. Front. Cell. Neurosci. 2018, 12, 74.
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104.
- Narayanan, S.V.; Dave, K.R.; Saul, I.; Perez-Pinzon, M.A. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke 2015, 46, 1626–1632.
- Yao, Y.; Miao, W.; Liu, Z.; Han, W.; Shi, K.; Shen, Y.; Li, H.; Liu, Q.; Fu, Y.; Huang, D.; et al. Dimethyl Fumarate and Monomethyl Fumarate Promote Post-Ischemic Recovery in Mice. Transl. Stroke Res. 2016, 7, 535–547.
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.R.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022.
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39.
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823.
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502.
- Oh, Y.S.; Jun, H.S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26.
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218.
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474.
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621.
- Culbreth, M.; Aschner, M. GSK-3beta, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000Research 2018, 7, 1043.
- Prema, A.; Janakiraman, U.; Manivasagam, T.; Thenmozhi, A.J. Neuroprotective effect of lycopene against MPTP induced experimental Parkinson’s disease in mice. Neurosci. Lett. 2015, 599, 12–19.
- Yang, C.H.; Yen, T.L.; Hsu, C.Y.; Thomas, P.A.; Sheu, J.R.; Jayakumar, T. Multi-Targeting Andrographolide, a Novel NF-kappaB Inhibitor, as a Potential Therapeutic Agent for Stroke. Int. J. Mol. Sci. 2017, 18, 1638.
- Huang, Y.; Mao, Y.; Li, H.; Shen, G.; Nan, G. Knockdown of Nrf2 inhibits angiogenesis by downregulating VEGF expression through PI3K/Akt signaling pathway in cerebral microvascular endothelial cells under hypoxic conditions. Biochem. Cell. Biol. 2018, 96, 475–482.
- Bai, J.; Yu, X.J.; Liu, K.L.; Wang, F.F.; Jing, G.X.; Li, H.B.; Zhang, Y.; Huo, C.J.; Li, X.; Gao, H.L.; et al. Central administration of tert-butylhydroquinone attenuates hypertension via regulating Nrf2 signaling in the hypothalamic paraventricular nucleus of hypertensive rats. Toxicol. Appl. Pharmacol. 2017, 333, 100–109.
- Li, M.T.; Ke, J.; Guo, S.F.; Wu, Y.; Bian, Y.F.; Shan, L.L.; Liu, Q.Y.; Huo, Y.J.; Guo, C.; Liu, M.Y.; et al. The Protective Effect of Quercetin on Endothelial Cells Injured by Hypoxia and Reoxygenation. Front. Pharmacol. 2021, 12, 732874.
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimer’s Dis. Rep. 2020, 4, 185–215.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
954
Revisions:
2 times
(View History)
Update Date:
27 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No