 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rocio Mora-Molina | -- | 2639 | 2022-09-22 17:01:31 | | | |
| 2 | Lindsay Dong | -54 word(s) | 2585 | 2022-09-26 06:01:25 | | |
Video Upload Options
The uncontrolled proliferation of malignant cells in growing tumors results in the generation of different stressors in the tumor microenvironment, such as nutrient shortage, hypoxia, and acidosis, among others, that disrupt endoplasmic reticulum (ER) homeostasis and may lead to ER stress. As a response to ER stress, both normal and tumor cells launch a set of signaling pathways known as the unfolded protein response (UPR) to restore ER proteostasis and maintain cell viability and function. However, under sustained ER stress, an apoptotic cell death process can be induced, although the role of the (TNF)-related apoptosis-inducing ligand receptor 2 (TRAIL-R2/DR5)-activated extrinsic pathway of apoptosis has not yet been thoroughly summarized.
1. Introduction
2. UPR Signaling Branches
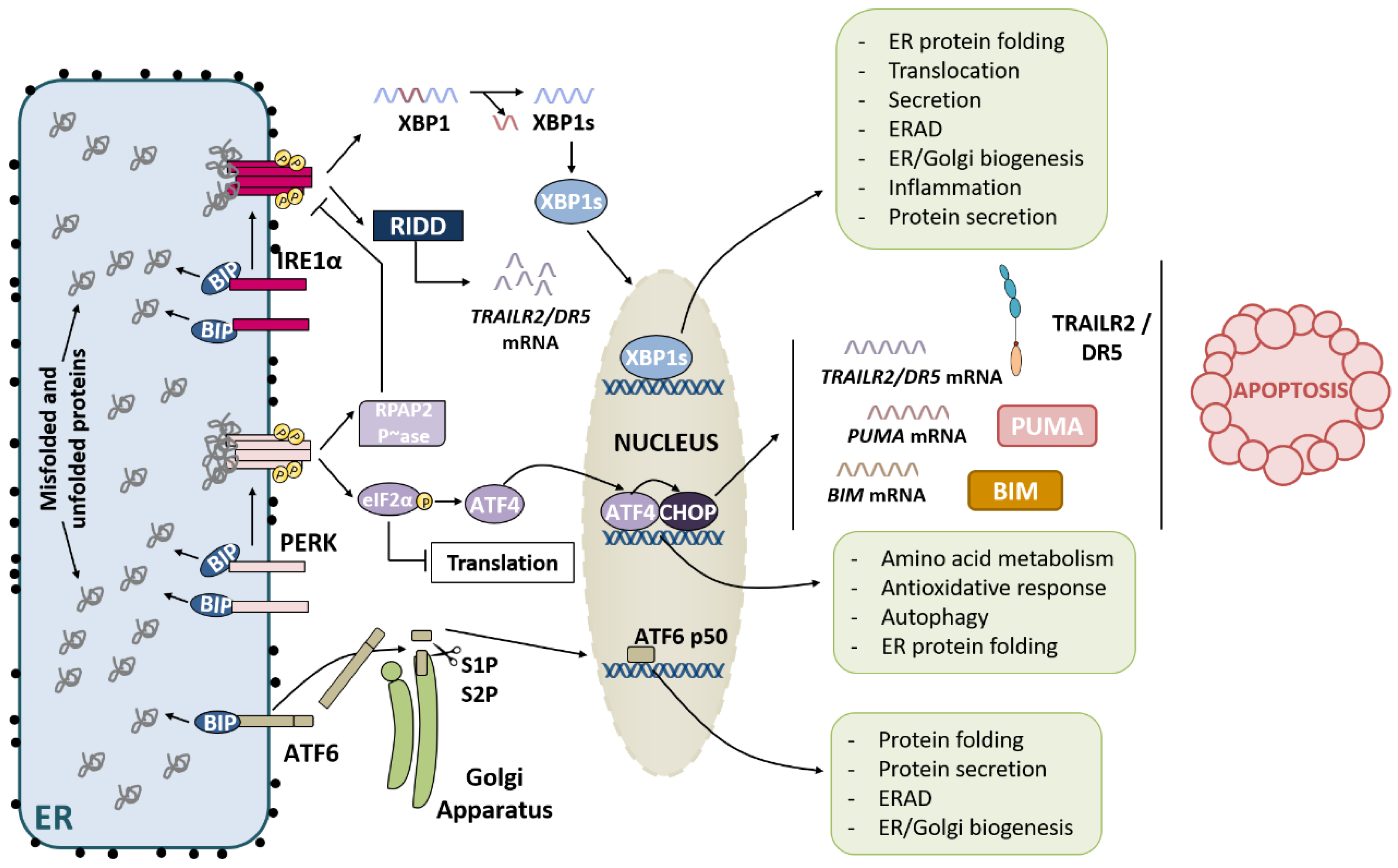
2.1. IRE1α Pathway
2.2. PERK Pathway
2.3. ATF6 Pathway
3. UPR Activation: Restore ER Homeostasis or Die in the Attempt
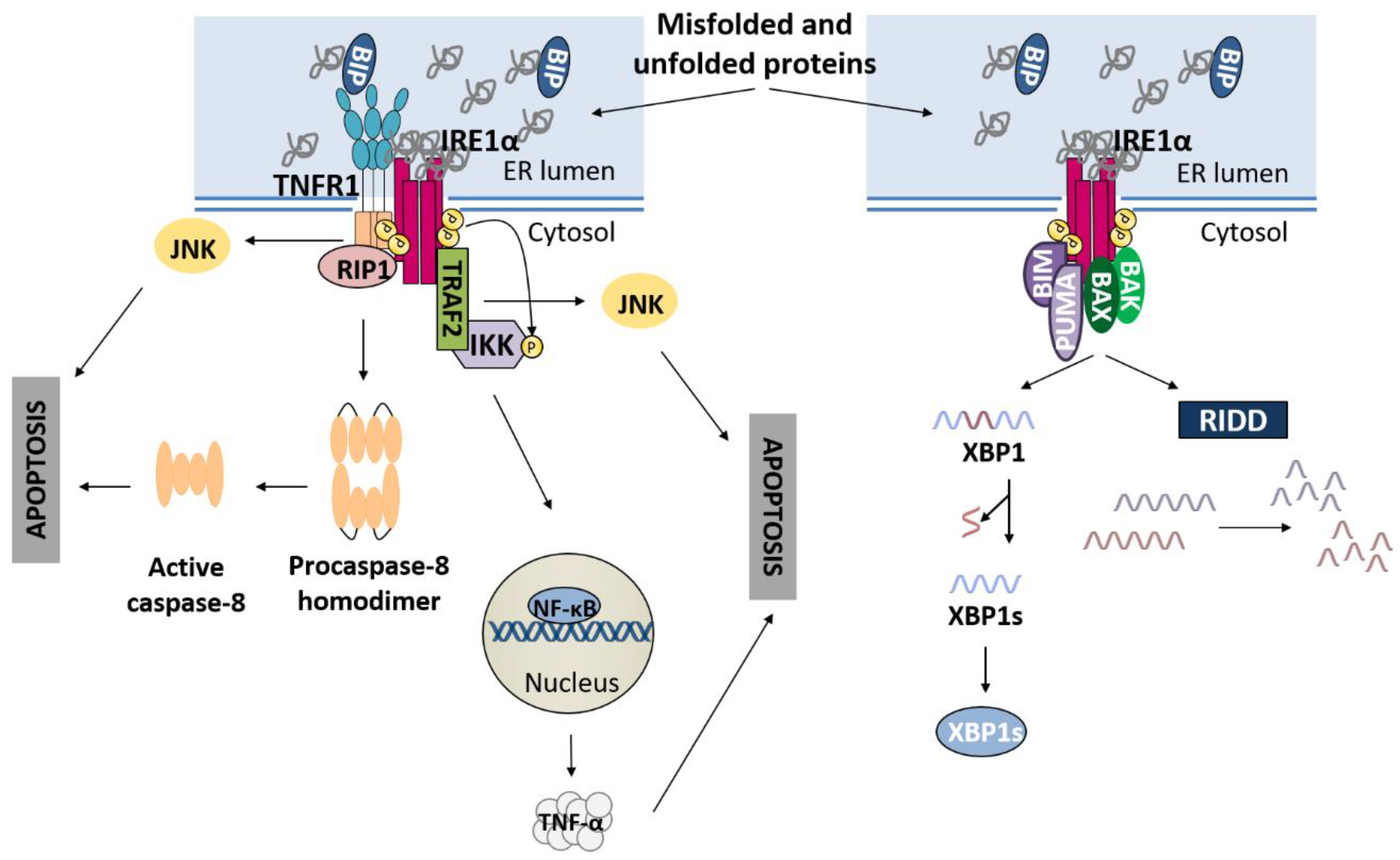
3.1. IRE1α Pathway and Apoptosis
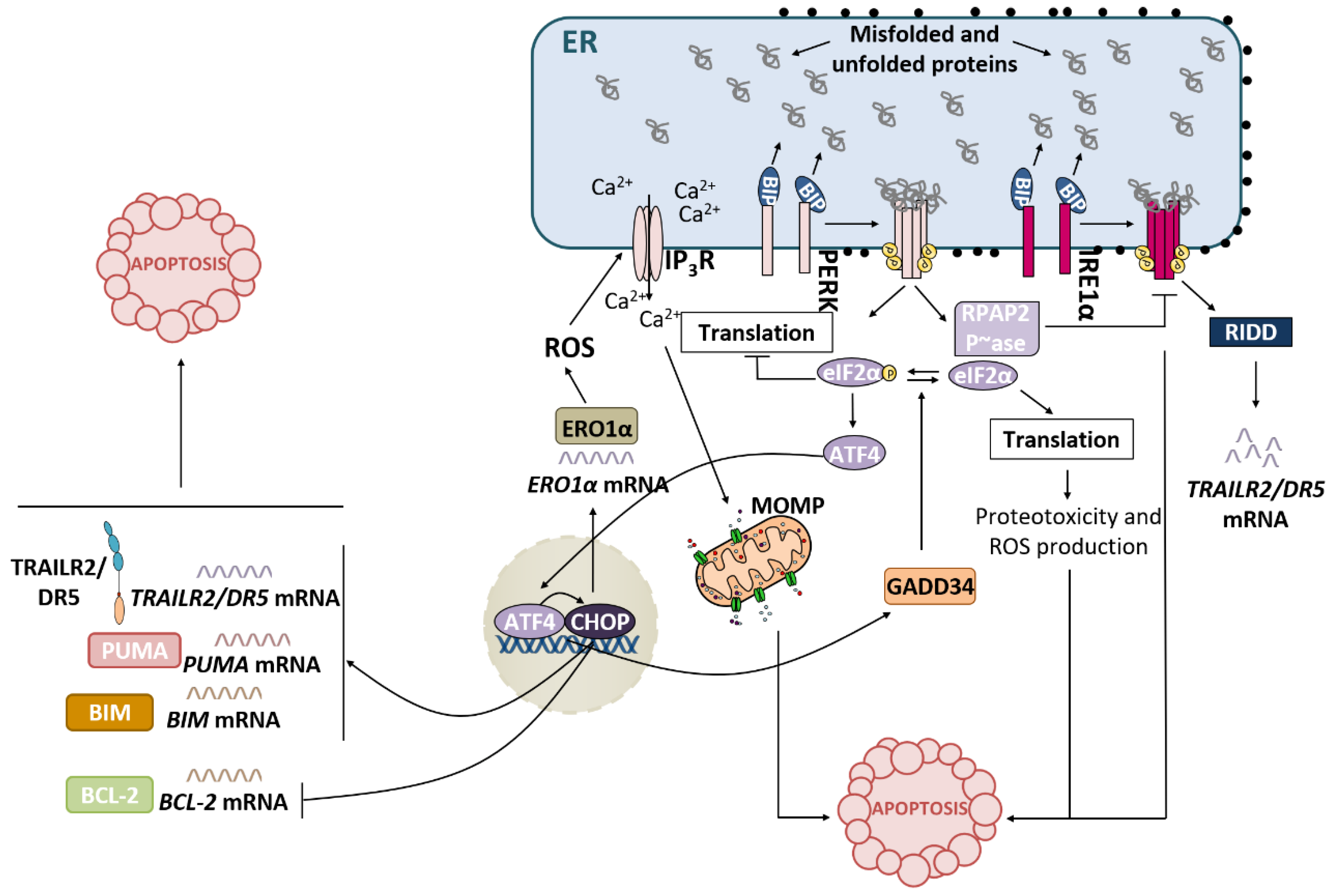
3.2. PERK Pathway and Apoptosis
Recently, the crosstalk between the PERK and IRE1α branches of the UPR has been described [40]. During the adaptive phase of the UPR, both pathways become activated to restore ER proteostasis. The endoribonuclease activity of IRE1α mediates RIDD of TRAILR2/DR5 mRNA, promoting cell survival. However, after prolonged ER stress, IRE1 signalling is attenuated, and the PERK pathway ultimately leads to apoptosis [41]. During the terminal phase of the UPR, RNA polymerase II-associated protein 2 (RPAP2) phosphatase acts downstream of PERK to attenuate IRE1 signalling, allowing TRAILR2/DR5 mRNA translation and the execution of apoptosis via the extrinsic pathway [40].
3.3. ATF6 Pathway and Apoptosis
4. Role of the TRAIL-R2-Activated Extrinsic Apoptotic Pathway in the Control of Tumor Progression
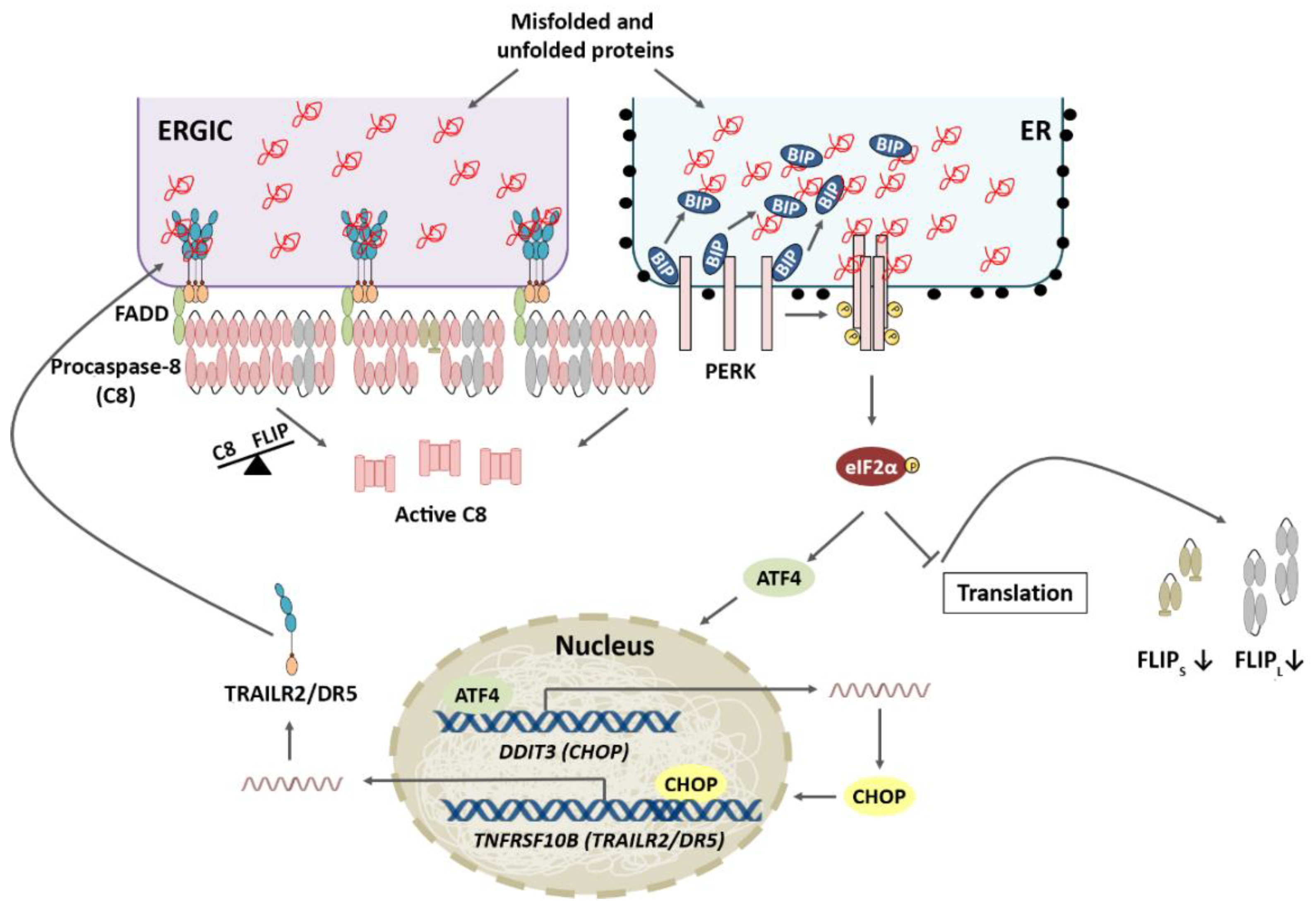
4.1. TRAIL-R2 Upregulation in Cells Undergoing ER Stress
4.2. Role of Cellular FLICE-like Inhibitory Protein (FLIP) in Apoptosis Regulation upon ER Stress
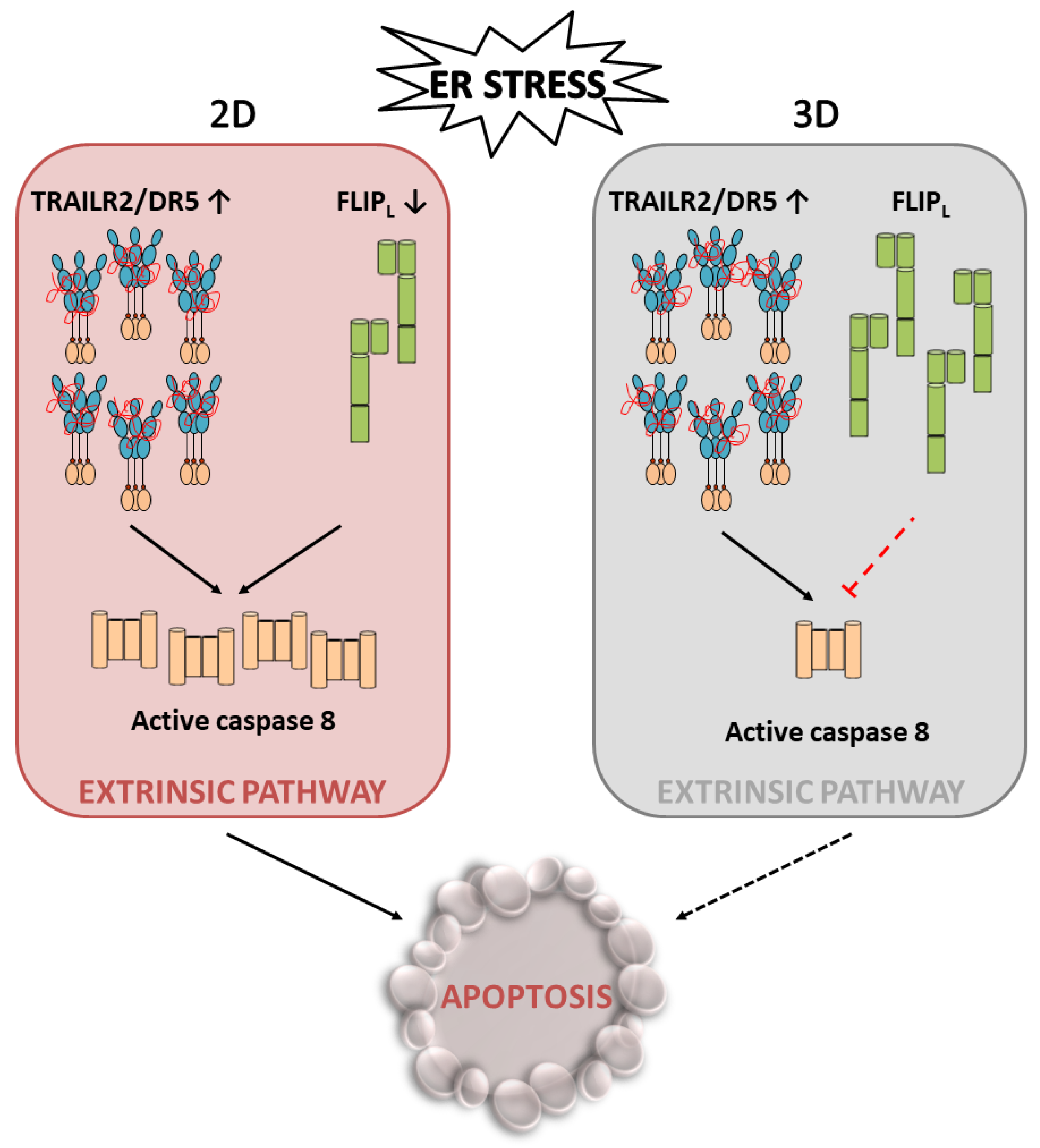
5. Conclusions
A high growth rate of cancer cells along with the poor vascularization of tumours result in stressful conditions in the tumour microenvironment, including a low oxygen supply and lack of nutrients, leading to metabolic stress. Metabolic stress can, in turn, adversely affect the environment of the endoplasmic reticulum (ER) and impact the maturation of nascent proteins. The resultant accumulation of the unfolded/misfolded proteins activates the UPR, which serves primarily to protect the cell during stress and helps restore homeostasis in the ER, facilitating tumour growth. However, if stress is prolonged or there is excessive stimulation of these signalling pathways, TRAILR2/DR5-mediated activation of the extrinsic apoptotic machinery and thereby cell death will occur. In this scenario, recent data suggest that cellular levels of FLIPL may play an important role in tumour cell fate decisions under the stressful conditions of the tumour microenvironment. Thus, in stressful situations, maintaining the levels of this protein that inhibits the extrinsic apoptosis pathway could enable the activation of an adaptive response in tumour cells and other tumour stromal cells, which would promote tumour growth and progression. Importantly, these results also reveal a dependence of tumour cells on maintaining FLIPL levels in the context of the tumour, which implies a vulnerability of these cells that could serve as a therapeutic target in cancer treatment.
References
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response. Dynamics and Metabolic Integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290.
- Wang, M.; Kaufman, R.J. Protein Misfolding in the Endoplasmic Reticulum as a Conduit to Human Disease. Nature 2016, 529, 326–335.
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181.
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438.
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364.
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A Link between the Unfolded Protein Response, Lipid Biosynthesis, and Biogenesis of the Endoplasmic Reticulum. J. Cell Biol. 2004, 167, 35–41.
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007, 27, 53–66.
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The EIF2α/ATF4 Pathway is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res. 2013, 41, 7683–7699.
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional Induction of Genes Encoding Endoplasmic Reticulum Resident Proteins Requires a Transmembrane Protein Kinase. Cell 1993, 73, 1197–1206.
- Wang, M.; Kaufman, R.J. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat. Rev. Cancer 2014, 14, 581–597.
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332.
- Karagöz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An Unfolded Protein-Induced Conformational Switch Activates Mammalian IRE1. eLife 2017, 6, e30700.
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891.
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96.
- Harding, H.P.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature 1999, 397, 271–274.
- Wang, P.; Li, J.; Tao, J.; Sha, B. The Luminal Domain of the ER Stress Sensor Protein PERK Binds Misfolded Proteins and Thereby Triggers PERK Oligomerization. J. Biol. Chem. 2018, 293, 4110–4121.
- Lu, P.D.; Harding, H.P.; Ron, D. Translation Reinitiation at Alternative Open Reading Frames Regulates Gene Expression in an Integrated Stress Response. J. Cell Biol. 2004, 167, 27–33.
- Vattem, K.M.; Wek, R.C. Reinitiation Involving Upstream ORFs Regulates ATF4 MRNA Translation in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2004, 10, 11269–11274.
- Wafa B’Chir; Anne-Catherine Maurin; Valérie Carraro; Julien Averous; Céline Jousse; Yuki Muranishi; Laurent Parry; Georges Stepien; Pierre Fafournoux; Alain Bruhat; et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Research 2013, 41, 7683-7699, 10.1093/nar/gkt563.
- Heather P. Harding; Yuhong Zhang; Huiquing Zeng; Isabel Novoa; Phoebe D. Lu; Marcella Calfon; Navid Sadri; Chi Yun; Brian Popko; Richard Paules; et al.David F. StojdlJohn C. BellThore HettmannJeffrey M. LeidenDavid Ron An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Molecular Cell 2003, 11, 619-633, 10.1016/s1097-2765(03)00105-9.
- Hirohito Yamaguchi; Hong-Gang Wang; CHOP Is Involved in Endoplasmic Reticulum Stress-induced Apoptosis by Enhancing DR5 Expression in Human Carcinoma Cells. Journal of Biological Chemistry 2004, 279, 45495-45502, 10.1074/jbc.m406933200.
- Karen D. McCullough; Jennifer L. Martindale; Lars-Oliver Klotz; Tak-Yee Aw; Nikki J. Holbrook; Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Molecular and Cellular Biology 2001, 21, 1249-1259, 10.1128/mcb.21.4.1249-1259.2001.
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799.
- Schindler, A.J.; Schekman, R. In Vitro Reconstitution of ER-Stress Induced ATF6 Transport in COPII Vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780.
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol. Cell 2000, 6, 1355–1364.
- Xi Chen; Juan R. Cubillos-Ruiz; Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nature Reviews Cancer 2020, 21, 71-88, 10.1038/s41568-020-00312-2.
- Miao Wang; Randal J. Kaufman; Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326-335, 10.1038/nature17041.
- Claudio Hetz; Feroz R. Papa; The Unfolded Protein Response and Cell Fate Control. Molecular Cell 2018, 69, 169-181, 10.1016/j.molcel.2017.06.017.
- Woehlbier, U.; Hetz, C. Modulating Stress Responses by the UPRosome: A Matter of Life and Death. Trends Biochem. Sci. 2011, 36, 329–337.
- Cano-González, A.; Mauro-Lizcano, M.; Iglesias-Serret, D.; Gil, J.; López-Rivas, A. Involvement of Both Caspase-8 and Noxa-Activated Pathways in Endoplasmic Reticulum Stress-Induced Apoptosis in Triple-Negative Breast Tumor Cells Article. Cell Death Dis. 2018, 9, 134.
- Martín-Pérez, R.; Palacios, C.; Yerbes, R.; Cano-González, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; López-Rivas, A. Activated ERBB2/HER2 Licenses Sensitivity to Apoptosis upon Endoplasmic Reticulum Stress through a PERK-Dependent Pathway. Cancer Res. 2014, 74, 1766–1777.
- Rodriguez, D.; Rojas-Rivera, D.; Hetz, C. Integrating Stress Signals at the Endoplasmic Reticulum: The BCL-2 Protein Family Rheostat. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 564–574.
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak Can Localize to the Endoplasmic Reticulum to Initiate Apoptosis. J. Cell Biol. 2003, 162, 59–69.
- Fumihiko Urano; Xiaozhong Wang; Anne Bertolotti; Yuhong Zhang; Peter Chung; Heather P. Harding; David Ron; Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664-666, 10.1126/science.287.5453.664.
- Qingfeng Yang; You‐Sun Kim; Yong Lin; Joseph Lewis; Len Neckers; Zheng‐Gang Liu; Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress‐induced activation of the MAP kinase JNK. EMBO reports 2006, 7, 622-627, 10.1038/sj.embor.7400687.
- Ping Hu; Zhang Han; Anthony D. Couvillon; Randal J. Kaufman; John H. Exton; Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway through IRE1α-Mediated NF-κB Activation and Down-Regulation of TRAF2 Expression. Molecular and Cellular Biology 2006, 26, 3071-3084, 10.1128/mcb.26.8.3071-3084.2006.
- Yann Estornes; Miguel Aguileta; Christel Dubuisson; Julie De Keyser; Vera Goossens; Kristof Kersse; Afshin Samali; Peter Vandenabeele; Mathieu Bertrand; RIPK1 promotes death receptor-independent caspase-8-mediated apoptosis under unresolved ER stress conditions. Cell Death & Disease 2014, 5, e1555-e1555, 10.1038/cddis.2014.523.
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cell Biol. 2013, 15, 481–490.
- Hery Urra; Estefanie Dufey; Fernanda Lisbona; Diego Rojas-Rivera; Claudio Hetz; When ER stress reaches a dead end. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2013, 1833, 3507-3517, 10.1016/j.bbamcr.2013.07.024.
- Tsun-Kai Chang; David A. Lawrence; Min Lu; Jenille Tan; Jonathan M. Harnoss; Scot A. Marsters; Peter Liu; Wendy Sandoval; Scott E. Martin; Avi Ashkenazi; et al. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Molecular Cell 2018, 71, 629-636.e5, 10.1016/j.molcel.2018.06.038.
- Min Lu; David A. Lawrence; Scot Marsters; Diego Acosta-Alvear; Philipp Kimmig; Aaron S. Mendez; Adrienne W. Paton; James C. Paton; Peter Walter; Avi Ashkenazi; et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98-101, 10.1126/science.1254312.
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the cis-Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2000, 20, 6755–6767.
- Yamaguchi, H.; Wang, H.G. CHOP is Involved in Endoplasmic Reticulum Stress-Induced Apoptosis by Enhancing DR5 Expression in Human Carcinoma Cells. J. Biol. Chem. 2004, 279, 45495–45502.
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101.
- Lam, M.; Marsters, S.; Ashkenazi, A.; Walter, P. Misfolded Proteins Bind and Activate Death Receptor 5 to Trigger Apoptosis during Unresolved Endoplasmic Reticulum Stress. eLife 2020, 9, e52291.
- Raffaella Iurlaro; Franziska Püschel; Clara Lucía León-Annicchiarico; Hazel O'Connor; Seamus J. Martin; Daniel Palou-Gramón; Estefanía Lucendo; Cristina Muñoz-Pinedo; Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Molecular and Cellular Biology 2017, 37, e00479-16, 10.1128/mcb.00479-16.
- Rosa Martín-Pérez; Rosario Yerbes; Rocío Mora-Molina; Ana Cano-González; Joaquin Arribas; Massimiliano Mazzone; Abelardo López-Rivas; Carmen Palacios; Oncogenic p95HER2/611CTF primes human breast epithelial cells for metabolic stress-induced down-regulation of FLIP and activation of TRAIL-R/Caspase-8-dependent apoptosis. Oncotarget 2017, 8, 93688-93703, 10.18632/oncotarget.21458.
- Day, T.W.; Sinn, A.L.; Huang, S.; Pollok, K.E.; Sandusky, G.E.; Safa, A.R. C-FLIP Gene Silencing Eliminates Tumor Cells in Breast Cancer Xenografts without Affecting Stromal Cells. Anticancer Res. 2009, 29, 3883–3886.
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-Operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849.
- Humphreys, L.M.; Fox, J.P.; Higgins, C.A.; Majkut, J.; Sessler, T.; McLaughlin, K.; McCann, C.; Roberts, J.Z.; Crawford, N.T.; McDade, S.S.; et al. A Revised Model of TRAIL -R2 DISC Assembly Explains How FLIP (L) Can Inhibit or Promote Apoptosis. EMBO Rep. 2020, 21, e49254.
- Palacios, C.; Yerbes, R.; López-Rivas, A. Flavopiridol Induces Cellular FLICE-Inhibitory Protein Degradation by the Proteasome and Promotes TRAIL-Induced Early Signaling and Apoptosis in Breast Tumor Cells. Cancer Res. 2006, 66, 8858–8869.
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.A.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. C-FLIP: A Key Regulator of Colorectal Cancer Cell Death. Cancer Res. 2007, 67, 5754–5762.
- Marini, E.S.; Giampietri, C.; Petrungaro, S.; Conti, S.; Filippini, A.; Scorrano, L.; Ziparo, E. The Endogenous Caspase-8 Inhibitor c-FLIPL Regulates ER Morphology and Crosstalk with Mitochondria. Cell Death Differ. 2015, 22, 1131–1143.
- Mora-Molina, R.; Stöhr, D.; Rehm, M.; López-Rivas, A. CFLIP Downregulation is an Early Event Required for Endoplasmic Reticulum Stress-Induced Apoptosis in Tumor Cells. Cell Death Dis. 2022, 13, 111.
- Millard, M.; Yakavets, I.; Zorin, V.; Kulmukhamedova, A.; Marchal, S.; Bezdetnaya, L. Drug Delivery to Solid Tumors: The Predictive Value of the Multicellular Tumor Spheroid Model for Nanomedicine Screening. Int. J. Nanomed. 2017, 12, 7993–8007.
- Nath, S.; Devi, G.R. Three-Dimensional Culture Systems in Cancer Research: Focus on Tumor Spheroid Model. Pharmacol. Ther. 2016, 163, 94–108.
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by CFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341.

