 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roberto Tonelli | -- | 3878 | 2022-09-23 10:39:45 | | | |
| 2 | Peter Tang | Meta information modification | 3878 | 2022-09-25 15:33:54 | | |
Video Upload Options
Among childhood cancers, neuroblastoma is the most diffuse solid tumor and the deadliest in children. While to date, the pathology has become progressively manageable with a significant increase in 5-year survival for its less aggressive form, high-risk neuroblastoma (HR-NB) remains a major issue with poor outcome and little survivability of patients. The staging system has also been improved to better fit patient needs and to administer therapies in a more focused manner in consideration of pathology features. New and improved therapies have been developed; nevertheless, low efficacy and high toxicity remain a staple feature of current high-risk neuroblastoma treatment. For this reason, more specific procedures are required, and new therapeutic targets are also needed for a precise medicine approach. In this scenario, MYCN is certainly one of the most interesting targets. Indeed, MYCN is one of the most relevant hallmarks of HR-NB, and many studies has been carried out in recent years to discover potent and specific inhibitors to block its activities and any related oncogenic function. N-Myc protein has been considered an undruggable target for a long time.
1. Introduction
|
INRGSS |
Age |
MYCN Amp |
SCA at 1 p or 11 q |
Ploidy |
INPC |
Differentiation |
Risk Group |
|---|---|---|---|---|---|---|---|
|
L1 |
No |
Any |
Any |
Any |
LR |
||
|
AMP |
LR or HR |
||||||
|
L2 |
<18 months |
No |
Absent |
DI > 1 |
FH |
IR |
|
|
Any |
Any |
Any |
IR |
||||
|
AMP |
Any |
Any |
Any |
HR |
|||
|
18 months–5 years |
No |
Any |
Any |
FH |
IR |
||
|
UH |
HR |
||||||
|
≥5 years |
No |
Any |
Any |
UH |
Differentiating |
IR |
|
|
Undifferentiated or poorly differentiated |
HR |
||||||
|
M |
<12 months |
No |
Any |
Any |
Any |
IR |
|
|
AMP |
HR |
||||||
|
12 to <18 months |
No |
Absent |
DI > 1 |
FH |
IR |
||
|
Present |
Any |
Any |
HR |
||||
|
Any |
DI = 1 |
Any |
HR |
||||
|
Any |
UH |
HR |
|||||
|
Any |
NA |
||||||
|
At least 1 feature |
Unfavorable |
HR |
|||||
|
AMP |
Any |
Any |
Any |
HR |
|||
|
≥18 months |
Any |
Any |
Any |
Any |
HR |
||
|
MS |
<12 months |
No bx |
No bx |
No bx |
No bx |
LR or IR |
|
|
No |
Absent |
DI > 1 |
FH |
LR or IR |
|||
|
Present |
Any |
Any |
IR |
||||
|
Any |
DI = 1 |
Any |
IR |
||||
|
Any |
UH |
IR |
|||||
|
AMP |
Any |
Any |
Any |
HR |
2. Diagnosis of High-Risk Neuroblastoma
2.1. Circulating Free DNA and Circulating Free Cells
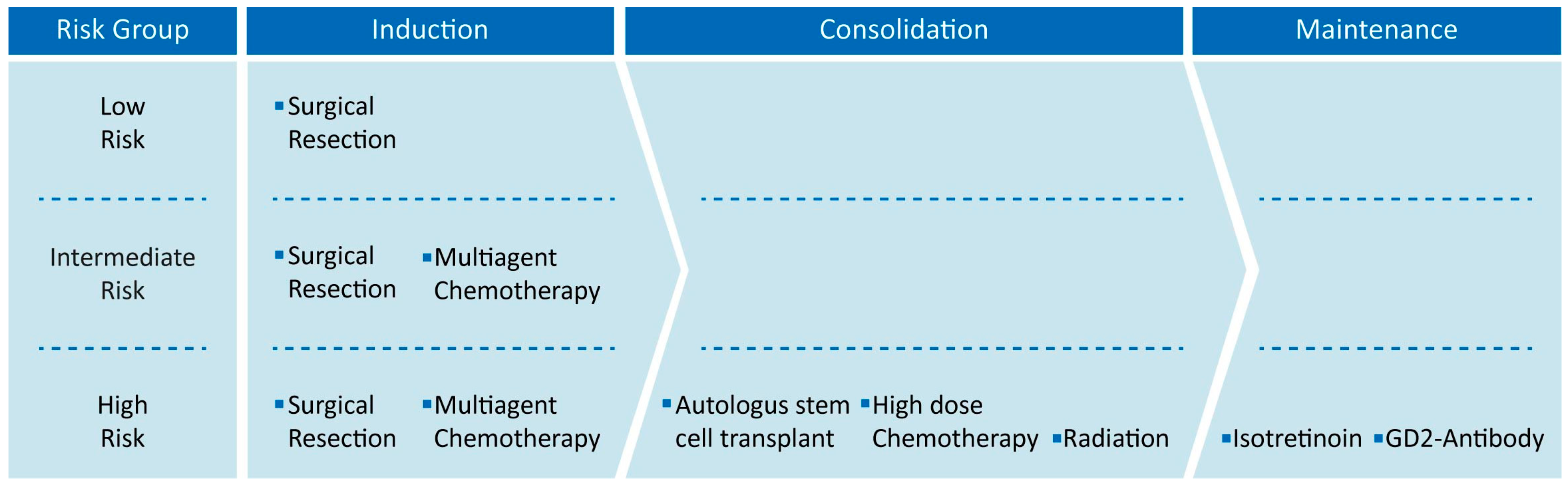
3. Current Therapies of High-Risk Neuroblastoma
4. MYCN as Prognostic Indicator in High-Risk Neuroblastoma
5. MYCN Determines High-Risk Neuroblastoma
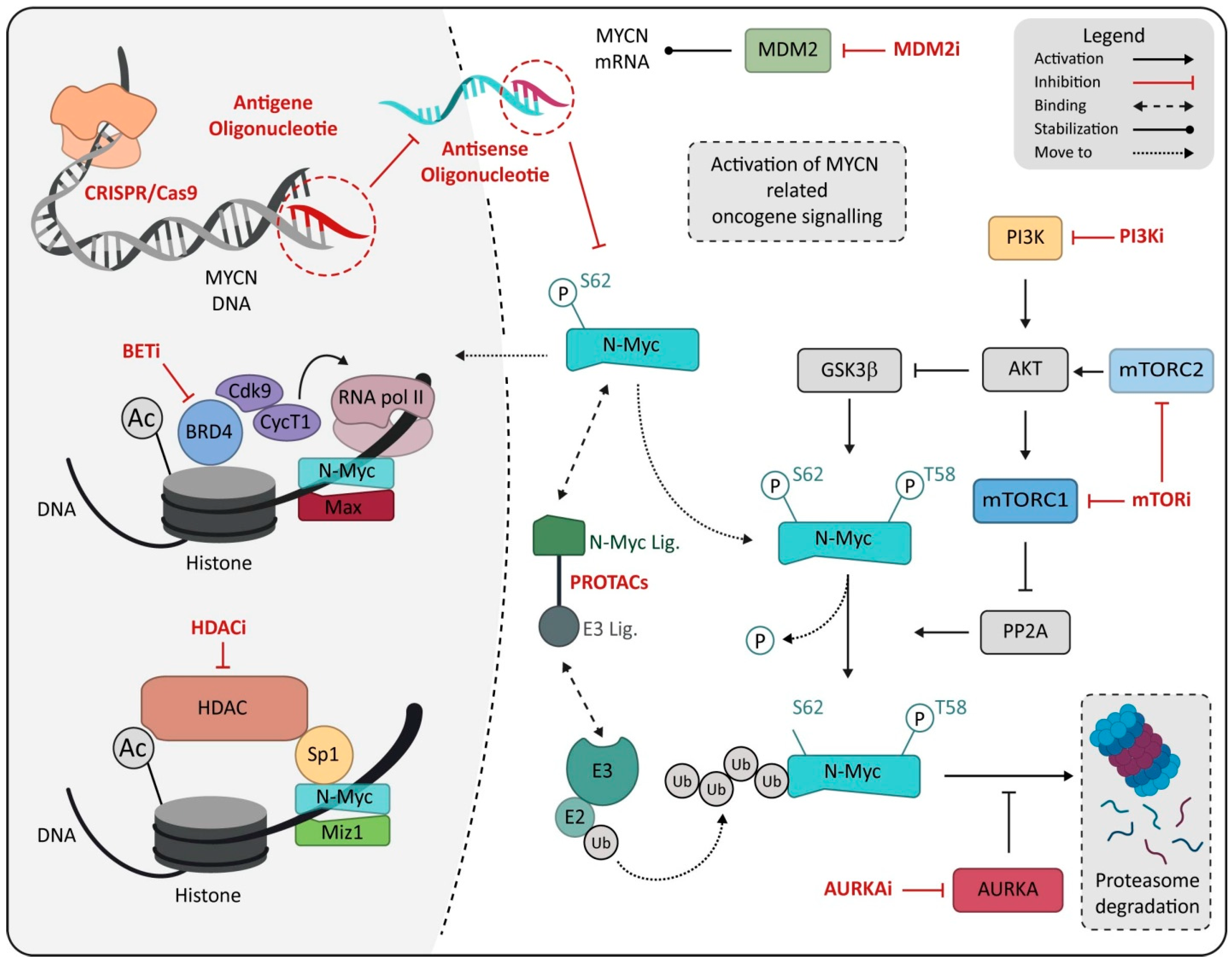
6. MYCN as Therapeutic Target
|
Therapy |
Therapeutic Strategy |
Availability |
|---|---|---|
|
Surgical Resection |
Tumor mass removal by surgical resection |
Standard Medical Practice |
|
Multimodal Chemotherapy |
Tumor cell elimination using non-specific chemical agents |
Standard Medical Practice |
|
Autologous stem cell transplantation |
Stem cell reinfusion after high dose chemotherapy |
Standard Medical Practice |
|
Radiation Therapy |
Tumor mass removal by radiations |
Standard Medical Practice |
|
Anti-GD2 Immunotherapy |
Induction of immune system stimulation |
Standard Medical Practice |
|
Isotretinoin |
Induction of tumor cell differentiation and proliferation arrest |
Standard Medical Practice |
|
BET Inhibitors |
Inhibition of specific molecular pathway related to MYCN |
Clinical Studies |
|
HDACs Inhibitors |
Inhibition of specific molecular pathway related to MYCN |
Clinical Studies |
|
PI3K/mTOR Inhibitors |
Inhibition of specific molecular pathway related to MYCN |
Clinical Studies |
|
Aurora Kinase-A Inhibitors |
Inhibition of specific molecular pathway related to MYCN |
Clinical Studies |
|
MDM2 inhibitors |
Inhibition of specific molecular pathway related to MYCN |
Clinical Studies |
|
MYCN direct inhibitor |
Specific MYCN expression inhibition or N-Myc protein degradation |
Preclinical Studies |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer Statistics, 2011: The Impact of Eliminating Socioeconomic and Racial Disparities on Premature Cancer Deaths. CA Cancer J. Clin. 2011, 61, 212–236.
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report from the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3229–3241.
- Ambros, P.F.; Ambros, I.M.; Brodeur, G.M.; Haber, M.; Khan, J.; Nakagawara, A.; Schleiermacher, G.; Speleman, F.; Spitz, R.; London, W.B.; et al. International Consensus for Neuroblastoma Molecular Diagnostics: Report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br. J. Cancer 2009, 100, 1471–1482.
- Pinto, N.; Mayfield, J.R.; Raca, G.; Applebaum, M.A.; Chlenski, A.; Sukhanova, M.; Bagatell, R.; Irwin, M.S.; Little, A.; Rawwas, J.; et al. Segmental Chromosomal Aberrations in Localized Neuroblastoma Can Be Detected in Formalin-Fixed Paraffin-Embedded Tissue Samples and are Associated with Recurrence: Segmental Chromosomal Aberrations in Localized Neuroblastoma. Pediatr. Blood Cancer 2016, 63, 1019–1023.
- Sokol, E.; Desai, A. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27.
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 289–297.
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415.
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120.
- Rubie, H.; Hartmann, O.; Michon, J.; Frappaz, D.; Coze, C.; Chastagner, P.; Baranzelli, M.C.; Plantaz, D.; Avet-Loiseau, H.; Bénard, J.; et al. N-Myc Gene Amplification is a Major Prognostic Factor in Localized Neuroblastoma: Results of the French NBL 90 Study. Neuroblastoma Study Group of the Société Francaise d’Oncologie Pédiatrique. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 1171–1182.
- Tang, X.X.; Zhao, H.; Kung, B.; Kim, D.Y.; Hicks, S.L.; Cohn, S.L.; Cheung, N.-K.; Seeger, R.C.; Evans, A.E.; Ikegaki, N. The MYCN Enigma: Significance of MYCN Expression in Neuroblastoma. Cancer Res. 2006, 66, 2826–2833.
- Nisen, P.D.; Waber, P.G.; Rich, M.A.; Pierce, S.; Garvin, J.R.; Gilbert, F.; Lanzkowsky, P. N-Myc Oncogene RNA Expression in Neuroblastoma. J. Natl. Cancer Inst. 1988, 80, 1633–1637.
- Jacobs, J.F.M.; van Bokhoven, H.; van Leeuwen, F.N.; Hulsbergen-van de Kaa, C.A.; de Vries, I.J.M.; Adema, G.J.; Hoogerbrugge, P.M.; de Brouwer, A.P.M. Regulation of MYCN Expression in Human Neuroblastoma Cells. BMC Cancer 2009, 9, 239.
- Scott, D.; Elsden, J.; Pearson, A.; Lunec, J. Genes Co-Amplified with MYCN in Neuroblastoma: Silent Passengers or Co-Determinants of Phenotype? Cancer Lett. 2003, 197, 81–86.
- Schwab, M. MYCN in Neuronal Tumours. Cancer Lett. 2004, 204, 179–187.
- Gherardi, S.; Valli, E.; Erriquez, D.; Perini, G. MYCN-Mediated Transcriptional Repression in Neuroblastoma: The Other Side of the Coin. Front. Oncol. 2013, 3, 42.
- Zafar, A.; Wang, W.; Liu, G.; Xian, W.; McKeon, F.; Zhou, J.; Zhang, R. Targeting the P53-MDM2 Pathway for Neuroblastoma Therapy: Rays of Hope. Cancer Lett. 2021, 496, 16–29.
- Wolpaw, A.J.; Bayliss, R.; Büchel, G.; Dang, C.V.; Eilers, M.; Gustafson, W.C.; Hansen, G.H.; Jura, N.; Knapp, S.; Lemmon, M.A.; et al. Drugging the “Undruggable” MYCN Oncogenic Transcription Factor: Overcoming Previous Obstacles to Impact Childhood Cancers. Cancer Res. 2021, 81, 1627–1632.
- Yue, Z.-X.; Huang, C.; Gao, C.; Xing, T.-Y.; Liu, S.-G.; Li, X.-J.; Zhao, Q.; Wang, X.-S.; Zhao, W.; Jin, M.; et al. MYCN Amplification Predicts Poor Prognosis Based on Interphase Fluorescence in Situ Hybridization Analysis of Bone Marrow Cells in Bone Marrow Metastases of Neuroblastoma. Cancer Cell Int. 2017, 17, 43.
- Campbell, K.; Gastier-Foster, J.M.; Mann, M.; Naranjo, A.H.; van Ryn, C.; Bagatell, R.; Matthay, K.K.; London, W.B.; Irwin, M.S.; Shimada, H.; et al. Association of MYCN Copy Number with Clinical Features, Tumor Biology, and Outcomes in Neuroblastoma: A Report from the Children’s Oncology Group. Cancer 2017, 123, 4224–4235.
- Swift, C.C.; Eklund, M.J.; Kraveka, J.M.; Alazraki, A.L. Updates in Diagnosis, Management, and Treatment of Neuroblastoma. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2018, 38, 566–580.
- Zhan, Y.; Shi, S.; Ehlerding, E.B.; Graves, S.A.; Goel, S.; Engle, J.W.; Liang, J.; Tian, J.; Cai, W. Radiolabeled, Antibody-Conjugated Manganese Oxide Nanoparticles for Tumor Vasculature Targeted Positron Emission Tomography and Magnetic Resonance Imaging. ACS Appl. Mater. Interfaces 2017, 9, 38304–38312.
- Bar-Sever, Z.; Biassoni, L.; Shulkin, B.; Kong, G.; Hofman, M.S.; Lopci, E.; Manea, I.; Koziorowski, J.; Castellani, R.; Boubaker, A.; et al. Guidelines on Nuclear Medicine Imaging in Neuroblastoma. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2009–2024.
- Kroiss, A.S. Current Status of Functional Imaging in Neuroblastoma, Pheochromocytoma, and Paraganglioma Disease. Wien. Med. Wochenschr. 2019, 169, 25–32.
- Sarioglu, F.C.; Salman, M.; Guleryuz, H.; Ozer, E.; Cecen, E.; Ince, D.; Olgun, N. Radiological Staging in Neuroblastoma: Computed Tomography or Magnetic Resonance Imaging? Pol. J. Radiol. 2019, 84, e46–e53.
- Sofka, C.M.; Semelka, R.C.; Kelekis, N.L.; Worawattanakul, S.; Chung, C.J.; Gold, S.; Fordham, L.A. Magnetic Resonance Imaging of Neuroblastoma Using Current Techniques. Magn. Reson. Imaging 1999, 17, 193–198.
- Wu, H.; Wu, C.; Zheng, H.; Wang, L.; Guan, W.; Duan, S.; Wang, D. Radiogenomics of Neuroblastoma in Pediatric Patients: CT-Based Radiomics Signature in Predicting MYCN Amplification. Eur. Radiol. 2021, 31, 3080–3089.
- Sharp, S.E.; Parisi, M.T.; Gelfand, M.J.; Yanik, G.A.; Shulkin, B.L. Functional-Metabolic Imaging of Neuroblastoma. Q. J. Nucl. Med. Mol. Imaging 2013, 57, 6–20.
- Campbell, K.; Shyr, D.; Bagatell, R.; Fischer, M.; Nakagawara, A.; Nieto, A.C.; Brodeur, G.M.; Matthay, K.K.; London, W.B.; DuBois, S.G. Comprehensive Evaluation of Context Dependence of the Prognostic Impact of MYCN Amplification in Neuroblastoma: A Report from the International Neuroblastoma Risk Group (INRG) Project. Pediatr. Blood Cancer 2019, 66, e27819.
- Yanishevski, D.; McCarville, M.B.; Doubrovin, M.; Spiegl, H.R.; Zhao, X.; Lu, Z.; Federico, S.M.; Furman, W.L.; Murphy, A.J.; Davidoff, A.M. Impact of MYCN Status on Response of High-Risk Neuroblastoma to Neoadjuvant Chemotherapy. J. Pediatr. Surg. 2020, 55, 130–134.
- Chan, H.S.; Gallie, B.L.; DeBoer, G.; Haddad, G.; Ikegaki, N.; Dimitroulakos, J.; Yeger, H.; Ling, V. MYCN Protein Expression as a Predictor of Neuroblastoma Prognosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 1699–1706.
- Van Heerden, J.; Esterhuizen, T.M.; Hendricks, M.; Poole, J.; Büchner, A.; Naidu, G.; du Plessis, J.; van Emmenes, B.; Uys, R.; Hadley, G.P.; et al. Age at Diagnosis as a Prognostic Factor in South African Children with Neuroblastoma. Pediatr. Blood Cancer 2021, 68, e28878.
- Sokol, E.; Desai, A.V.; Applebaum, M.A.; Valteau-Couanet, D.; Park, J.R.; Pearson, A.D.J.; Schleiermacher, G.; Irwin, M.S.; Hogarty, M.; Naranjo, A.; et al. Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index are Independently Prognostic in Neuroblastoma: An INRG Project. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1906–1918.
- Mathew, P.; Valentine, M.B.; Bowman, L.C.; Rowe, S.T.; Nash, M.B.; Valentine, V.A.; Cohn, S.L.; Castleberry, R.P.; Brodeur, G.M.; Look, A.T. Detection of MYCN Gene Amplification in Neuroblastoma by Fluorescence in Situ Hybridization: A Pediatric Oncology Group Study. Neoplasia 2001, 3, 105–109.
- Squire, J.A.; Thorner, P.A.; Marrano, P.A.; Parkinson, D.I.; Ng, Y.K.; Gerrie, B.L.; Chilton-Macneill, S.; Zielenska, M. Identification of MYCN Copy Number Heterogeneity by Direct FISH Analysis of Neuroblastoma Preparations. Mol. Diagn. 1996, 1, 281–289.
- Marrano, P.; Irwin, M.S.; Thorner, P.S. Heterogeneity of MYCN Amplification in Neuroblastoma at Diagnosis, Treatment, Relapse, and Metastasis. Genes Chromosom. Cancer 2017, 56, 28–41.
- Marrugo-Ramírez, J.; Mir, M.; Samitier, J. Blood-Based Cancer Biomarkers in Liquid Biopsy: A Promising Non-Invasive Alternative to Tissue Biopsy. Int. J. Mol. Sci. 2018, 19, 2877.
- Namløs, H.M.; Boye, K.; Mishkin, S.J.; Barøy, T.; Lorenz, S.; Bjerkehagen, B.; Stratford, E.W.; Munthe, E.; Kudlow, B.A.; Myklebost, O.; et al. Noninvasive Detection of CtDNA Reveals Intratumor Heterogeneity and is Associated with Tumor Burden in Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2018, 17, 2473–2480.
- Combaret, V.; Audoynaud, C.; Iacono, I.; Favrot, M.-C.; Schell, M.; Bergeron, C.; Puisieux, A. Circulating MYCN DNA as a Tumor-Specific Marker in Neuroblastoma Patients. Cancer Res. 2002, 62, 3646–3648.
- Combaret, V.; Bergeron, C.; Noguera, R.; Iacono, I.; Puisieux, A. Circulating MYCN DNA Predicts MYCN-Amplification in Neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 8919–8920; author reply 8920.
- Trigg, R.M.; Turner, S.D.; Shaw, J.A.; Jahangiri, L. Diagnostic Accuracy of Circulating-Free DNA for the Determination of MYCN Amplification Status in Advanced-Stage Neuroblastoma: A Systematic Review and Meta-Analysis. Br. J. Cancer 2020, 122, 1077–1084.
- Gotoh, T.; Hosoi, H.; Iehara, T.; Kuwahara, Y.; Osone, S.; Tsuchiya, K.; Ohira, M.; Nakagawara, A.; Kuroda, H.; Sugimoto, T. Prediction of MYCN Amplification in Neuroblastoma Using Serum DNA and Real-Time Quantitative Polymerase Chain Reaction. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 5205–5210.
- Iehara, T.; Yagyu, S.; Gotoh, T.; Ouchi, K.; Yoshida, H.; Miyachi, M.; Kikuchi, K.; Sugimoto, T.; Hosoi, H. A Prospective Evaluation of Liquid Biopsy for Detecting MYCN Amplification in Neuroblastoma Patients. Jpn. J. Clin. Oncol. 2019, 49, 743–748.
- Pinzani, P.; D’Argenio, V.; del Re, M.; Pellegrini, C.; Cucchiara, F.; Salvianti, F.; Galbiati, S. Updates on Liquid Biopsy: Current Trends and Future Perspectives for Clinical Application in Solid Tumors. Clin. Chem. Lab. Med. 2021, 59, 1181–1200.
- Rifatbegovic, F.; Frech, C.; Abbasi, M.R.; Taschner-Mandl, S.; Weiss, T.; Schmidt, W.M.; Schmidt, I.; Ladenstein, R.; Ambros, I.M.; Ambros, P.F. Neuroblastoma Cells Undergo Transcriptomic Alterations upon Dissemination into the Bone Marrow and Subsequent Tumor Progression. Int. J. Cancer 2018, 142, 297–307.
- Reza, K.K.; Dey, S.; Wuethrich, A.; Wang, J.; Behren, A.; Antaw, F.; Wang, Y.; Sina, A.A.I.; Trau, M. In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment. ACS Nano 2021, 15, 11231–11243.
- Lodrini, M.; Wünschel, J.; Thole-Kliesch, T.M.; Grimaldi, M.; Sprüssel, A.; Linke, R.B.; Hollander, J.F.; Tiburtius, D.; Künkele, A.; Schulte, J.H.; et al. Circulating Cell-Free DNA Assessment in Biofluids from Children with Neuroblastoma Demonstrates Feasibility and Potential for Minimally Invasive Molecular Diagnostics. Cancers 2022, 14, 2080.
- Beltran, H.; Jendrisak, A.; Landers, M.; Mosquera, J.M.; Kossai, M.; Louw, J.; Krupa, R.; Graf, R.P.; Schreiber, N.A.; Nanus, D.M.; et al. The Initial Detection and Partial Characterization of Circulating Tumor Cells in Neuroendocrine Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1510–1519.
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation Analysis of Cell-Free DNA and Single Circulating Tumor Cells in Metastatic Breast Cancer Patients with High Circulating Tumor Cell Counts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 88–96.
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Children 2018, 5, 114.
- DuBois, S.G.; Macy, M.E.; Henderson, T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 768–780.
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and Biological Approach to Risk Stratification and Treatment. Cell Tissue Res. 2018, 372, 195–209.
- Baker, D.L.; Schmidt, M.L.; Cohn, S.L.; Maris, J.M.; London, W.B.; Buxton, A.; Stram, D.; Castleberry, R.P.; Shimada, H.; Sandler, A.; et al. Outcome after Reduced Chemotherapy for Intermediate-Risk Neuroblastoma. N. Engl. J. Med. 2010, 363, 1313–1323.
- Rubie, H.; de Bernardi, B.; Gerrard, M.; Canete, A.; Ladenstein, R.; Couturier, J.; Ambros, P.; Munzer, C.; Pearson, A.D.J.; Garaventa, A.; et al. Excellent Outcome with Reduced Treatment in Infants with Nonmetastatic and Unresectable Neuroblastoma without MYCN Amplification: Results of the Prospective INES 99.1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 449–455.
- Strother, D.R.; London, W.B.; Schmidt, M.L.; Brodeur, G.M.; Shimada, H.; Thorner, P.; Collins, M.H.; Tagge, E.; Adkins, S.; Reynolds, C.P.; et al. Outcome after Surgery Alone or with Restricted Use of Chemotherapy for Patients with Low-Risk Neuroblastoma: Results of Children’s Oncology Group Study P9641. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1842–1848.
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of High-Risk Neuroblastoma with Intensive Chemotherapy, Radiotherapy, Autologous Bone Marrow Transplantation, and 13-Cis-Retinoic Acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173.
- Yanik, G.; Naranjo, A.; Parisi, M.T.; Shulkin, B.L.; Nadel, H.; Gelfand, M.J.; Ladenstein, R.; Boubaker, A.; Poetschger, U.; Valteau-Couanet, D.; et al. Impact of Post-Induction Curie Scores in High-Risk Neuroblastoma. Biol. Blood Marrow Transplant. 2015, 21, S107.
- Yanik, G.A.; Parisi, M.T.; Naranjo, A.; Nadel, H.; Gelfand, M.J.; Park, J.R.; Ladenstein, R.L.; Poetschger, U.; Boubaker, A.; Valteau-Couanet, D.; et al. Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children’s Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 502–508.
- Park, J.R.; Kreissman, S.G.; London, W.B.; Naranjo, A.; Cohn, S.L.; Hogarty, M.D.; Tenney, S.C.; Haas-Kogan, D.; Shaw, P.J.; Kraveka, J.M.; et al. Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients with High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 2019, 322, 746–755.
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and Recent Advances in the Treatment of Neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386.
- Ozkaynak, M.F.; Gilman, A.L.; London, W.B.; Naranjo, A.; Diccianni, M.B.; Tenney, S.C.; Smith, M.; Messer, K.S.; Seeger, R.; Reynolds, C.P.; et al. A Comprehensive Safety Trial of Chimeric Antibody 14.18 With GM-CSF, IL-2, and Isotretinoin in High-Risk Neuroblastoma Patients Following Myeloablative Therapy: Children’s Oncology Group Study ANBL0931. Front. Immunol. 2018, 9, 1355.
- Tonini, G.P.; Boni, L.; Pession, A.; Rogers, D.; Iolascon, A.; Basso, G.; Cordero di Montezemolo, L.; Casale, F.; Pession, A.; Perri, P.; et al. MYCN Oncogene Amplification in Neuroblastoma is Associated with Worse Prognosis, except in Stage 4s: The Italian Experience with 295 Children. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 85–93.
- Brodeur, G.M. Neuroblastoma: Biological Insights into a Clinical Enigma. Nat. Rev. Cancer 2003, 3, 203–216.
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-Myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124.
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N-Myc Oncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116.
- Lampis, S.; Raieli, S.; Montemurro, L.; Bartolucci, D.; Amadesi, C.; Bortolotti, S.; Angelucci, S.; Scardovi, A.L.; Nieddu, G.; Cerisoli, L.; et al. The MYCN Inhibitor BGA002 Restores the Retinoic Acid Response Leading to Differentiation or Apoptosis by the MTOR Block in MYCN-Amplified Neuroblastoma. J. Exp. Clin. Cancer Res. 2022, 41, 160.
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC–Driven Tumors. Cancer Discov. 2018, 8, 150–163.
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell Cycle Control across the Eukaryotic Kingdom. Trends Cell Biol. 2013, 23, 345–356.
- Woo, C.-W.; Tan, F.; Cassano, H.; Lee, J.; Lee, K.C.; Thiele, C.J. Use of RNA Interference to Elucidate the Effect of MYCN on Cell Cycle in Neuroblastoma. Pediatr. Blood Cancer 2008, 50, 208–212.
- Cage, T.A.; Chanthery, Y.; Chesler, L.; Grimmer, M.; Knight, Z.; Shokat, K.; Weiss, W.A.; Gustafson, W.C. Downregulation of MYCN through PI3K Inhibition in Mouse Models of Pediatric Neural Cancer. Front. Oncol. 2015, 5, 111.
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K Inhibition Results in Enhanced Estrogen Receptor Function and Dependence in Hormone Receptor-Positive Breast Cancer. Sci. Transl. Med. 2015, 7, 283ra51.
- Bouchard, C.; Dittrich, O.; Kiermaier, A.; Dohmann, K.; Menkel, A.; Eilers, M.; Lüscher, B. Regulation of Cyclin D2 Gene Expression by the Myc/Max/Mad Network: Myc-Dependent TRRAP Recruitment and Histone Acetylation at the Cyclin D2 Promoter. Genes Dev. 2001, 15, 2042–2047.
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F Integrates Cell Cycle Progression with DNA Repair, Replication, and G(2)/M Checkpoints. Genes Dev. 2002, 16, 245–256.
- Lasorella, A.; Stegmüller, J.; Guardavaccaro, D.; Liu, G.; Carro, M.S.; Rothschild, G.; de la Torre-Ubieta, L.; Pagano, M.; Bonni, A.; Iavarone, A. Degradation of Id2 by the Anaphase-Promoting Complex Couples Cell Cycle Exit and Axonal Growth. Nature 2006, 442, 471–474.
- Kuzyk, A.; Gartner, J.; Mai, S. Identification of Neuroblastoma Subgroups Based on Three-Dimensional Telomere Organization. Transl. Oncol. 2016, 9, 348–356.
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT Rearrangements are Frequent in Neuroblastoma and Identify Aggressive Tumors. Nat. Genet. 2015, 47, 1411–1414.
- Ham, J.; Costa, C.; Sano, R.; Lochmann, T.L.; Sennott, E.M.; Patel, N.U.; Dastur, A.; Gomez-Caraballo, M.; Krytska, K.; Hata, A.N.; et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer Cell 2016, 29, 159–172.
- Chen, L.; Iraci, N.; Gherardi, S.; Gamble, L.D.; Wood, K.M.; Perini, G.; Lunec, J.; Tweddle, D.A. P53 is a Direct Transcriptional Target of MYCN in Neuroblastoma. Cancer Res. 2010, 70, 1377–1388.
- Hou, H.; Sun, D.; Zhang, X. The Role of MDM2 Amplification and Overexpression in Therapeutic Resistance of Malignant Tumors. Cancer Cell Int. 2019, 19, 216.
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A Threshold Mechanism Mediates P53 Cell Fate Decision between Growth Arrest and Apoptosis. Cell Death Differ. 2013, 20, 576–588.
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8.
- Barbieri, E.; Mehta, P.; Chen, Z.; Zhang, L.; Slack, A.; Berg, S.; Shohet, J.M. MDM2 Inhibition Sensitizes Neuroblastoma to Chemotherapy-Induced Apoptotic Cell Death. Mol. Cancer Ther. 2006, 5, 2358–2365.
- Yogev, O.; Barker, K.; Sikka, A.; Almeida, G.S.; Hallsworth, A.; Smith, L.M.; Jamin, Y.; Ruddle, R.; Koers, A.; Webber, H.T.; et al. P53 Loss in MYC-Driven Neuroblastoma Leads to Metabolic Adaptations Supporting Radioresistance. Cancer Res. 2016, 76, 3025–3035.
- Qi, D.-L.; Cobrinik, D. MDM2 but Not MDM4 Promotes Retinoblastoma Cell Proliferation through P53-Independent Regulation of MYCN Translation. Oncogene 2017, 36, 1760–1769.
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer 2011, 11, 85–95.
- Diers, A.R.; Broniowska, K.A.; Chang, C.-F.; Hogg, N. Pyruvate Fuels Mitochondrial Respiration and Proliferation of Breast Cancer Cells: Effect of Monocarboxylate Transporter Inhibition. Biochem. J. 2012, 444, 561–571.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033.
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232.
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerström, L.; Westermark, U.K.; Larsson, K.; Munksgaard Persson, M.; Hultenby, K.; Lehtiö, J.; Einvik, C.; et al. MYC Inhibition Induces Metabolic Changes Leading to Accumulation of Lipid Droplets in Tumor Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263.
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350.
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012, 22, 631–644.
- Wahlström, T.; Henriksson, M.A. Impact of MYC in Regulation of Tumor Cell Metabolism. Biochim. Biophys. Acta 2015, 1849, 563–569.
- Tao, L.; Mohammad, M.A.; Milazzo, G.; Moreno-Smith, M.; Patel, T.D.; Zorman, B.; Badachhape, A.; Hernandez, B.E.; Wolf, A.B.; Zeng, Z.; et al. MYCN-Driven Fatty Acid Uptake is a Metabolic Vulnerability in Neuroblastoma. Nat. Commun. 2022, 13, 3728.
- Alborzinia, H.; Flórez, A.F.; Kreth, S.; Brückner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN Mediates Cysteine Addiction and Sensitizes Neuroblastoma to Ferroptosis. Nat. Cancer 2022, 3, 471–485.
- Montemurro, L.; Raieli, S.; Angelucci, S.; Bartolucci, D.; Amadesi, C.; Lampis, S.; Scardovi, A.L.; Venturelli, L.; Nieddu, G.; Cerisoli, L.; et al. A Novel MYCN-Specific Antigene Oligonucleotide Deregulates Mitochondria and Inhibits Tumor Growth in MYCN-Amplified Neuroblastoma. Cancer Res. 2019, 79, 6166–6177.
- Vaughan, L.; Clarke, P.A.; Barker, K.; Chanthery, Y.; Gustafson, C.W.; Tucker, E.; Renshaw, J.; Raynaud, F.; Li, X.; Burke, R.; et al. Inhibition of MTOR-Kinase Destabilizes MYCN and is a Potential Therapy for MYCN-Dependent Tumors. Oncotarget 2016, 7, 57525–57544.
- Schramm, A.; Köster, J.; Marschall, T.; Martin, M.; Schwermer, M.; Fielitz, K.; Büchel, G.; Barann, M.; Esser, D.; Rosenstiel, P.; et al. Next-Generation RNA Sequencing Reveals Differential Expression of MYCN Target Genes and Suggests the MTOR Pathway as a Promising Therapy Target in MYCN-Amplified Neuroblastoma. Int. J. Cancer 2013, 132, E106–E115.
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832.
- Raieli, S.; di Renzo, D.; Lampis, S.; Amadesi, C.; Montemurro, L.; Pession, A.; Hrelia, P.; Fischer, M.; Tonelli, R. MYCN Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front. Oncol. 2021, 11, 625207.
- Nallasamy, P.; Chava, S.; Verma, S.S.; Mishra, S.; Gorantla, S.; Coulter, D.W.; Byrareddy, S.N.; Batra, S.K.; Gupta, S.C.; Challagundla, K.B. PD-L1, Inflammation, Non-Coding RNAs, and Neuroblastoma: Immuno-Oncology Perspective. Semin. Cancer Biol. 2018, 52, 53–65.
- Melaiu, O.; Mina, M.; Chierici, M.; Boldrini, R.; Jurman, G.; Romania, P.; D’Alicandro, V.; Benedetti, M.C.; Castellano, A.; Liu, T.; et al. PD-L1 is a Therapeutic Target of the Bromodomain Inhibitor JQ1 and, Combined with HLA Class I, a Promising Prognostic Biomarker in Neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4462–4472.
- Noguera, R.; Fredlund, E.; Piqueras, M.; Pietras, A.; Beckman, S.; Navarro, S.; Påhlman, S. HIF-1α and HIF-2α are Differentially Regulated In Vivo in Neuroblastoma: High HIF-1α Correlates Negatively to Advanced Clinical Stage and Tumor Vascularization. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7130–7136.
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e8.
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1364–1370.
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of Programmed Death-Ligand 1 Expression and Tumor-Associated Immune Cells in Pediatric Cancer Tissues. Cancer 2017, 123, 3807–3815.
- Hahn, M.; Glass, T.; Koke, J. Extracellular Matrix Effects on a Neuroblastoma Cell Line. Cytobios 2000, 102, 7–19.
- Meyer, A.; van Golen, C.M.; Kim, B.; van Golen, K.L.; Feldman, E.L. Integrin Expression Regulates Neuroblastoma Attachment and Migration. Neoplasia 2004, 6, 332–342.
- Young, S.A.; McCabe, K.E.; Bartakova, A.; Delaney, J.; Pizzo, D.P.; Newbury, R.O.; Varner, J.A.; Schlaepfer, D.D.; Stupack, D.G. Integrin A4 Enhances Metastasis and May Be Associated with Poor Prognosis in MYCN-Low Neuroblastoma. PLoS ONE 2015, 10, e0120815.
- Erdreich-Epstein, A.; Shimada, H.; Groshen, S.; Liu, M.; Metelitsa, L.S.; Kim, K.S.; Stins, M.F.; Seeger, R.C.; Durden, D.L. Integrins Alpha(v)Beta3 and Alpha(v)Beta5 are Expressed by Endothelium of High-Risk Neuroblastoma and Their Inhibition is Associated with Increased Endogenous Ceramide. Cancer Res. 2000, 60, 712–721.
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253.
- Willumsen, N.; Thomsen, L.B.; Bager, C.L.; Jensen, C.; Karsdal, M.A. Quantification of Altered Tissue Turnover in a Liquid Biopsy: A Proposed Precision Medicine Tool to Assess Chronic Inflammation and Desmoplasia Associated with a pro-Cancerous Niche and Response to Immuno-Therapeutic Anti-Tumor Modalities. Cancer Immunol. Immunother. 2018, 67, 1–12.
- Ng, M.R.; Brugge, J.S. A Stiff Blow from the Stroma: Collagen Crosslinking Drives Tumor Progression. Cancer Cell 2009, 16, 455–457.
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan Inhibits Collagen I Synthesis and Improves the Distribution and Efficacy of Nanotherapeutics in Tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914.
- Sugiura, Y.; Shimada, H.; Seeger, R.C.; Laug, W.E.; DeClerck, Y.A. Matrix Metalloproteinases-2 and -9 are Expressed in Human Neuroblastoma: Contribution of Stromal Cells to Their Production and Correlation with Metastasis. Cancer Res. 1998, 58, 2209–2216.
- Sans-Fons, M.G.; Sole, S.; Sanfeliu, C.; Planas, A.M. Matrix Metalloproteinase-9 and Cell Division in Neuroblastoma Cells and Bone Marrow Macrophages. Am. J. Pathol. 2010, 177, 2870–2885.
- Ara, T.; Fukuzawa, M.; Kusafuka, T.; Komoto, Y.; Oue, T.; Inoue, M.; Okada, A. Immunohistochemical Expression of MMP-2, MMP-9, and TIMP-2 in Neuroblastoma: Association with Tumor Progression and Clinical Outcome. J. Pediatr. Surg. 1998, 33, 1272–1278.
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410.
- Ramani, P.; Nash, R.; Radevsky, L.; Patel, A.; Luckett, M.; Rogers, C. VEGF-C, VEGF-D and VEGFR-3 Expression in Peripheral Neuroblastic Tumours. Histopathology 2012, 61, 1006–1016.
- Chlenski, A.; Liu, S.; Cohn, S.L. The Regulation of Angiogenesis in Neuroblastoma. Cancer Lett. 2003, 197, 47–52.
- Meitar, D.; Crawford, S.E.; Rademaker, A.W.; Cohn, S.L. Tumor Angiogenesis Correlates with Metastatic Disease, N-Myc Amplification, and Poor Outcome in Human Neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1996, 14, 405–414.
- Ribatti, D.; Vacca, A.; Nico, B.; de Falco, G.; Giuseppe Montaldo, P.; Ponzoni, M. Angiogenesis and Anti-Angiogenesis in Neuroblastoma. Eur. J. Cancer 2002, 38, 750–757.
- Rössler, J.; Taylor, M.; Geoerger, B.; Farace, F.; Lagodny, J.; Peschka-Süss, R.; Niemeyer, C.M.; Vassal, G. Angiogenesis as a Target in Neuroblastoma. Eur. J. Cancer 2008, 44, 1645–1656.
- Chanthery, Y.H.; Gustafson, W.C.; Itsara, M.; Persson, A.; Hackett, C.S.; Grimmer, M.; Charron, E.; Yakovenko, S.; Kim, G.; Matthay, K.K.; et al. Paracrine Signaling through MYCN Enhances Tumor-Vascular Interactions in Neuroblastoma. Sci. Transl. Med. 2012, 4, 115ra3.
- Kang, J.; Rychahou, P.G.; Ishola, T.A.; Mourot, J.M.; Evers, B.M.; Chung, D.H. N-Myc is a Novel Regulator of PI3K-Mediated VEGF Expression in Neuroblastoma. Oncogene 2008, 27, 3999–4007.
- Singh, A.R.; Joshi, S.; Burgoyne, A.M.; Sicklick, J.K.; Ikeda, S.; Kono, Y.; Garlich, J.R.; Morales, G.A.; Durden, D.L. Single Agent and Synergistic Activity of the “First-in-Class” Dual PI3K/BRD4 Inhibitor SF1126 with Sorafenib in Hepatocellular Carcinoma. Mol. Cancer Ther. 2016, 15, 2553–2562.
- Joshi, S.; Singh, A.R.; Durden, D.L. Pan-PI-3 Kinase Inhibitor SF1126 Shows Antitumor and Antiangiogenic Activity in Renal Cell Carcinoma. Cancer Chemother. Pharmacol. 2015, 75, 595–608.
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A Macrophage-Dominant PI3K Isoform Controls Hypoxia-Induced HIF1α and HIF2α Stability and Tumor Growth, Angiogenesis, and Metastasis. Mol. Cancer Res. 2014, 12, 1520–1531.
- Singh, A.R.; Joshi, S.; George, E.; Durden, D.L. Anti-Tumor Effect of a Novel PI3-Kinase Inhibitor, SF1126, in (12) V-Ha-Ras Transgenic Mouse Glioma Model. Cancer Cell Int. 2014, 14, 105.
- Xing, F.; Saidou, J.; Watabe, K. Cancer Associated Fibroblasts (CAFs) in Tumor Microenvironment. Front. Biosci. Landmark Ed. 2010, 15, 166–179.
- Yazhou, C.; Wenlv, S.; Weidong, Z.; Licun, W. Clinicopathological Significance of Stromal Myofibroblasts in Invasive Ductal Carcinoma of the Breast. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2004, 25, 290–295.
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive Stroma in Human Prostate Cancer: Induction of Myofibroblast Phenotype and Extracellular Matrix Remodeling. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2912–2923.
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348.
- Silzle, T.; Kreutz, M.; Dobler, M.A.; Brockhoff, G.; Knuechel, R.; Kunz-Schughart, L.A. Tumor-Associated Fibroblasts Recruit Blood Monocytes into Tumor Tissue. Eur. J. Immunol. 2003, 33, 1311–1320.
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-Beta Signalling Inhibitors for Cancer Therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022.
- Hashimoto, O.; Yoshida, M.; Koma, Y.-I.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of Cancer-Associated Fibroblasts and Tumour-Associated Macrophages for Neuroblastoma Development. J. Pathol. 2016, 240, 211–223.
- Kakarla, S.; Song, X.-T.; Gottschalk, S. Cancer-Associated Fibroblasts as Targets for Immunotherapy. Immunotherapy 2012, 4, 1129–1138.
- Fakhrai, H.; Dorigo, O.; Shawler, D.L.; Lin, H.; Mercola, D.; Black, K.L.; Royston, I.; Sobol, R.E. Eradication of Established Intracranial Rat Gliomas by Transforming Growth Factor Beta Antisense Gene Therapy. Proc. Natl. Acad. Sci. USA 1996, 93, 2909–2914.
- Braoudaki, M.; Hatziagapiou, K.; Zaravinos, A.; Lambrou, G.I. MYCN in Neuroblastoma: “Old Wine into New Wineskins”. Diseases 2021, 9, 78.
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233.
- Buechner, J.; Einvik, C. N-Myc and Noncoding RNAs in Neuroblastoma. Mol. Cancer Res. 2012, 10, 1243–1253.
- Beckers, A.; van Peer, G.; Carter, D.R.; Mets, E.; Althoff, K.; Cheung, B.B.; Schulte, J.H.; Mestdagh, P.; Vandesompele, J.; Marshall, G.M.; et al. MYCN-Targeting MiRNAs are Predominantly Downregulated during MYCN-driven Neuroblastoma Tumor Formation. Oncotarget 2015, 6, 5204–5216.
- Molenaar, J.J.; Domingo-Fernández, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B Induces Neuroblastoma and Enhances MYCN Levels via Let-7 Suppression. Nat. Genet. 2012, 44, 1199–1206.
- Powers, J.T.; Tsanov, K.M.; Pearson, D.S.; Roels, F.; Spina, C.S.; Ebright, R.; Seligson, M.; de Soysa, Y.; Cahan, P.; Theißen, J.; et al. Multiple Mechanisms Disrupt the Let-7 MicroRNA Family in Neuroblastoma. Nature 2016, 535, 246–251.
- Misiak, D.; Hagemann, S.; Bell, J.L.; Busch, B.; Lederer, M.; Bley, N.; Schulte, J.H.; Hüttelmaier, S. The MicroRNA Landscape of MYCN-Amplified Neuroblastoma. Front. Oncol. 2021, 11, 647737.
- Mogilyansky, E.; Rigoutsos, I. The MiR-17/92 Cluster: A Comprehensive Update on Its Genomics, Genetics, Functions and Increasingly Important and Numerous Roles in Health and Disease. Cell Death Differ. 2013, 20, 1603–1614.
- Mestdagh, P.; Boström, A.-K.; Impens, F.; Fredlund, E.; van Peer, G.; de Antonellis, P.; von Stedingk, K.; Ghesquière, B.; Schulte, S.; Dews, M.; et al. The MiR-17-92 MicroRNA Cluster Regulates Multiple Components of the TGF-β Pathway in Neuroblastoma. Mol. Cell 2010, 40, 762–773.
- Armstrong, B.C.; Krystal, G.W. Isolation and Characterization of Complementary DNA for N-Cym, a Gene Encoded by the DNA Strand Opposite to N-Myc. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1992, 3, 385–390.
- Vadie, N.; Saayman, S.; Lenox, A.; Ackley, A.; Clemson, M.; Burdach, J.; Hart, J.; Vogt, P.K.; Morris, K.V. MYCNOS Functions as an Antisense RNA Regulating MYCN. RNA Biol. 2015, 12, 893–899.
- Suenaga, Y.; Islam, S.M.R.; Alagu, J.; Kaneko, Y.; Kato, M.; Tanaka, Y.; Kawana, H.; Hossain, S.; Matsumoto, D.; Yamamoto, M.; et al. NCYM, a Cis-Antisense Gene of MYCN, Encodes a de Novo Evolved Protein That Inhibits GSK3β Resulting in the Stabilization of MYCN in Human Neuroblastomas. PLoS Genet. 2014, 10, e1003996.
- O’Brien, E.M.; Selfe, J.L.; Martins, A.S.; Walters, Z.S.; Shipley, J.M. The Long Non-Coding RNA MYCNOS-01 Regulates MYCN Protein Levels and Affects Growth of MYCN-Amplified Rhabdomyosarcoma and Neuroblastoma Cells. BMC Cancer 2018, 18, 217.
- Liu, P.Y.; Atmadibrata, B.; Mondal, S.; Tee, A.E.; Liu, T. NCYM is Upregulated by LncUSMycN and Modulates N-Myc Expression. Int. J. Oncol. 2016, 49, 2464–2470.
- Decock, A.; Ongenaert, M.; Vandesompele, J.; Speleman, F. Neuroblastoma Epigenetics: From Candidate Gene Approaches to Genome-Wide Screenings. Epigenetics 2011, 6, 962–970.
- Westerlund, I.; Shi, Y.; Toskas, K.; Fell, S.M.; Li, S.; Surova, O.; Södersten, E.; Kogner, P.; Nyman, U.; Schlisio, S.; et al. Combined Epigenetic and Differentiation-Based Treatment Inhibits Neuroblastoma Tumor Growth and Links HIF2α to Tumor Suppression. Proc. Natl. Acad. Sci. USA 2017, 114, E6137–E6146.
- Parodi, F.; Carosio, R.; Ragusa, M.; di Pietro, C.; Maugeri, M.; Barbagallo, D.; Sallustio, F.; Allemanni, G.; Pistillo, M.P.; Casciano, I.; et al. Epigenetic Dysregulation in Neuroblastoma: A Tale of MiRNAs and DNA Methylation. Biochim. Biophys. Acta 2016, 1859, 1502–1514.
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular Pathogenesis and Therapy. Annu. Rev. Med. 2015, 66, 49–63.
- Pession, A.; Tonelli, R. The MYCN Oncogene as a Specific and Selective Drug Target for Peripheral and Central Nervous System Tumors. Curr. Cancer Drug Targets 2005, 5, 273–283.
- Fletcher, J.I.; Ziegler, D.S.; Trahair, T.N.; Marshall, G.M.; Haber, M.; Norris, M.D. Too Many Targets, Not Enough Patients: Rethinking Neuroblastoma Clinical Trials. Nat. Rev. Cancer 2018, 18, 389–400.
- Andresen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.H.; Lundström, P.; et al. Transient Structure and Dynamics in the Disordered C-Myc Transactivation Domain Affect Bin1 Binding. Nucleic Acids Res. 2012, 40, 6353–6366.
- Bayliss, R.; Burgess, S.G.; Leen, E.; Richards, M.W. A Moving Target: Structure and Disorder in Pursuit of Myc Inhibitors. Biochem. Soc. Trans. 2017, 45, 709–717.
- Kohl, N.E.; Legouy, E.; DePinho, R.A.; Nisen, P.D.; Smith, R.K.; Gee, C.E.; Alt, F.W. Human N-Myc is Closely Related in Organization and Nucleotide Sequence to c-Myc. Nature 1986, 319, 73–77.
- Esposito, M.R.; Aveic, S.; Seydel, A.; Tonini, G.P. Neuroblastoma Treatment in the Post-Genomic Era. J. Biomed. Sci. 2017, 24, 14.


